- Home
- GEP
- Genomes
- Genome Browser
- Tools
- My Data
- Help
bigWig Track Format
The bigWig format is useful for dense, continuous data that will be displayed in the Genome
Browser as a graph. BigWig files are created from wiggle (wig)
type files using the program wigToBigWig. Alternatively, bigWig files can be created
from bedGraph files, using the program
bedGraphToBigWig.
The bigWig files are in an indexed binary format. The main advantage of this format is that only those portions of the file needed to display a particular region are transferred to the Genome Browser server. Because of this, bigWig files have considerably faster display performance than regular wiggle files when working with large data sets. The bigWig file remains on your local web-accessible server (http, https or ftp), not on the UCSC server, and only the portion needed for the currently displayed chromosomal position is locally cached as a "sparse file". If you do not have access to a web-accessible server and need hosting space for your bigWig files, please see the Hosting section of the Track Hub Help documentation.
Wiggle data must be continuous and consist of equally sized elements. If your data is sparse or
contains elements of varying sizes, use the bedGraph format instead of
the wiggle format. If you have a very large bedGraph data set, you can convert it to the bigWig
format using the bedGraphToBigWig program. For details, see Example
#3 below. Refer to
this wiki page for help in selecting the graphing track data format most
appropriate for the type of data you have.
Note that the wigToBigWig utility uses a substantial amount of memory: approximately
50% more memory than that of the uncompressed wiggle input file. While running the
wigToBigWig utility, we recommend that you monitor the system's memory usage
with the top command. The bedGraphToBigWig utility uses about 25%
more RAM than the uncompressed bedGraph input file.
Creating a bigWig track
To create a bigWig track from a wiggle file, follow these steps:
Step 1. Create a wig format file following the directions here. When converting a wig file to a bigWig file, you are limited to one track of data in your input file; therefore, you must create a separate wig file for each data track.
Step 2. Remove any existing "track" or "browser" lines from your wig file so that it contains only data.
Step 3.
Download the wigToBigWig program from the
binary utilities directory.
Step 4.
Use the fetchChromSizes script from the
same directory to create the
chrom.sizes file for the UCSC database with which you are working (e.g., hg19).
If the assembly genNom is hosted by UCSC, chrom.sizes can be a URL like:
http://hgdownload.soe.ucsc.edu/goldenPath/genNom/bigZips/genNom.chrom.sizes
Step 5.
Use the wigToBigWig utility to create the bigWig file from your wig file:
wigToBigWig input.wig chrom.sizes myBigWig.bwNote that the wigToBigWig program also accepts gzipped wig input files.
Step 6. Move the newly created bigWig file (myBigWig.bw) to a web-accessible http, https, or ftp location.
Step 7. If the file name ends with a .bigWig or .bw suffix, you can paste the URL directly into the custom track management page, click "submit" and view the file as a track in the Genome Browser. By default, the file name will be used to name the track. To configure the track label or other visualization options, you must create a track line, as shown in Step 8.
Step 8. Construct a custom track using a single track line. The most basic version of the track line will look something like this:
track type=bigWig name="My Big Wig" description="A Graph of Data from My Lab" bigDataUrl=http://myorg.edu/mylab/myBigWig.bwPaste the custom track line into the text box on the custom track management page.
bigWig custom track lines can have several optional parameters, including:
autoScale <on|off> # default is on alwaysZero <on|off> # default is off gridDefault <on|off> # default is off maxHeightPixels <max:default:min> # default is 128:128:11 graphType <bar|points> # default is bar viewLimits <lower:upper> # default is range found in data viewLimitsMax <lower:upper> # suggested bounds of viewLimits, but not enforced yLineMark <real-value> # default is 0.0 yLineOnOff <on|off> # default is off windowingFunction <mean+whiskers|maximum|mean|minimum> # default is maximum, mean+whiskers is recommended smoothingWindow <off|[2-16]> # default is off transformFunc <NONE|LOG> # default is NONE
For further information on custom bigWig track settings, see the Track Database Definition Document. For more information on how bigWig settings are used in native Genome Browser tracks, see the Configuring graph-based tracks page.
Examples
Example #1
In this example, you create a bigWig custom track using an existing bigWig file on the UCSC http server. The file contains data that spans chromosome 21 on the hg19 assembly.
To create a custom track using this bigWig file:
-
Paste the URL
http://genome.ucsc.edu/goldenPath/help/examples/bigWigExample.bwonto the custom track management page for the human assembly hg19 (Feb. 2009). - Click the "submit" button.
- On the next page that displays, click the "chr21" link in the custom track listing to view the bigWig track at position chr21:33,031,597-33,041,570 in the Genome Browser.
Alternatively, you can customize the track display by including track and browser lines that define certain parameters:
-
Construct a track line that references the bigWigExample.bw file:
track type=bigWig name="Example One" description="A bigWig file" bigDataUrl=http://genome.ucsc.edu/goldenPath/help/examples/bigWigExample.bw -
Include the following browser line to ensure that the custom track opens at the correct
position:
browser position chr21:33,031,597-33,041,570 - Paste the browser and track lines onto the custom track management page for the human assembly hg19 (Feb. 2009), click the "submit" button, then click the "chr21" link in the custom track listing to view the bigWig track in the Genome Browser.
Example #2
In this example, you will create your own bigWig file from an existing wiggle file.
- Save this wiggle file to your computer (Steps 1 and 2 in Creating a bigWig format track, above).
- Save the file hg19.chrom.sizes to your computer. This file contains the chrom.sizes for the human (hg19) assembly (Step 4, above).
-
Download the
wigToBigWigutility (step 3, above). -
Run the utility to create the bigWig output file (step 5, above):
wigToBigWig wigVarStepExample.gz hg19.chrom.sizes myBigWig.bw - Place the newly created bigWig file (myBigWig.bw) on a web-accessible server (step 6, above).
- Paste the URL of the bigWig file into the custom track entry form, or construct a track line that points to your bigWig file (step 7, above).
- Create the custom track on the human assembly hg19 (Feb. 2009), and view it in the Genome Browser (step 8, above). Note that the original wiggle file spans only chromosome 21.
Example #3
To create a bigWig track from a bedGraph file, follow these steps:
- Create a bedGraph format file following the directions here. When converting a bedGraph file to a bigWig file, you are limited to one track of data in your input file; therefore, you must create a separate bedGraph file for each data track.
- Remove any existing track or browser lines from your bedGraph file so that it contains only data.
-
Download the
bedGraphToBigWigprogram from the binary utilities directory. -
Use the
fetchChromSizesscript from the same directory to create the chrom.sizes file for the UCSC database with which you are working (e.g., hg19). If the assemblygenNomis hosted by UCSC, chrom.sizes can be a URL likehttp://hgdownload.soe.ucsc.edu/goldenPath/genNom/bigZips/genNom.chrom.sizes -
Use the
bedGraphToBigWigutility to create a bigWig file from your bedGraph file:
(Note that the bedGraphToBigWig program DOES NOT accept gzipped bedGraph input files.)bedGraphToBigWig in.bedGraph chrom.sizes myBigWig.bw -
Move the newly created bigWig file (
myBigWig.bw) to a web-accessible http, https, or ftp location. - Paste the URL into the custom track entry form or construct a custom track using a single track line.
- Paste the custom track line into the text box on the custom track management page.
Example #4
In this example, we will display a bigWig with a dynamic sequence logo using logo=on.
-
Construct a track line that references the bigWigExample.bw file:
track type=bigWig logo=on name="Example dynseq" description="A bigWig file with logo=on dynseq" visibility=full autoScale=off bigDataUrl=http://genome.ucsc.edu/goldenPath/help/examples/bigWigExample.bw -
Include the following browser line to ensure that the custom track opens at the correct
position:
browser position chr21:33,037,000-33,037,050 - Paste the browser and track lines onto the custom track management page for the human assembly hg19 (Feb. 2009), click the "submit" button, then click the "chr21" link in the custom track listing to view the bigWig track in the Genome Browser.
You can also load the above by clicking this link.
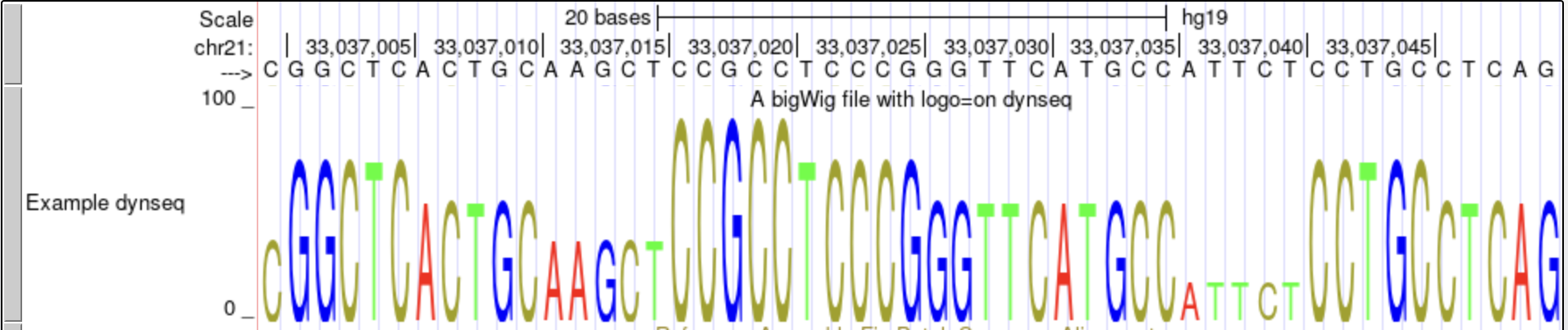
This dynseq display scales nucleotide characters by user-specified, base-resolution scores and was developed by the Kundaje Lab.
Sharing your data with others
If you would like to share your bigWig data track with a colleague, learn how to create a URL by looking at Example #6 on this page.
Extracting data from the bigWig format
Because bigWig files are indexed binary files, it can be difficult to extract data from them. UCSC has developed the following programs to assist in working with these files, available from the binary utilities directory.
-
bigWigToBedGraph— converts a bigWig file to ASCII bedGraph format. -
bigWigToWig— converts a bigWig file to wig format. Note: if a bigWig file was created from a bedGraph, bigWigToWig will revert the file back to bedGraph. -
bigWigSummary— extracts summary information from a bigWig file. -
bigWigAverageOverBed— computes the average score of a bigWig over each bed, which may have introns. -
bigWigInfo— prints out information about a bigWig file.
These utilities accept either file path names or URLs to files as input. As with all UCSC Genome Browser programs, simply type the program name (with no parameters) on the command line to view the usage statement.
In some cases, bigWigSummary and bigWigAverageOverBed will produce very
similar results, but in other cases, the results may differ. This is due to data-handling
differences between the two programs. Summary levels are used with bigWigSummary;
therefore, some rounding errors and border conditions are encountered when extracting data over
relatively small regions. In contrast, the bigWigAverageOverBed utility uses the actual
data, which ensures the highest level of accuracy.