- Home
- GEP
- Genomes
- Genome Browser
- Tools
- My Data
- Help
Genome Browser User Guide
Contents
What does the Genome Browser do?
Configuring the Genome Browser display
Annotation track descriptions
Using BLAT alignments
Getting Started with:
DNA text formatting
Converting data between assemblies
Downloading genome data
Creating and managing custom annotation tracks
Getting started on Track Hubs
Track Hubs in a single file
Using the VisiGene Image Browser
Browse the Genome Browser mailing list.
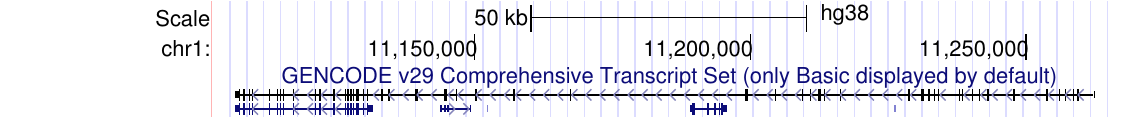
What does the Genome Browser do?
As vertebrate genome sequences near completion and research re-focuses on their analysis, the issue of effective sequence display becomes critical: it is not helpful to have 3 billion letters of genomic DNA shown as plain text! As an alternative, the UCSC Genome Browser provides a rapid and reliable display of any requested portion of genomes at any scale, together with dozens of aligned annotation tracks (known genes, predicted genes, ESTs, mRNAs, CpG islands, assembly gaps and coverage, chromosomal bands, mouse homologies, and more). Half of the annotation tracks are computed at UCSC from publicly available sequence data. The remaining tracks are provided by collaborators worldwide. Users can also add their own custom tracks to the browser for educational or research purposes.
The Genome Browser stacks annotation tracks beneath genome coordinate positions, allowing rapid visual correlation of different types of information. The user can look at a whole chromosome to get a feel for gene density, open a specific cytogenetic band to see a positionally mapped disease gene candidate, or zoom in to a particular gene to view its spliced ESTs and possible alternative splicing. The Genome Browser itself does not draw conclusions; rather, it collates all relevant information in one location, leaving the exploration and interpretation to the user.
The Genome Browser supports text and sequence based searches that provide quick, precise access to any region of specific interest. Secondary links from individual entries within annotation tracks lead to sequence details and supplementary off-site databases. To control information overload, tracks need not be displayed in full. Tracks can be hidden, collapsed into a condensed or single-line display, or filtered according to the user's criteria. Zooming and scrolling controls help to narrow or broaden the displayed chromosomal range to focus on the exact region of interest. Clicking on an individual item within a track opens a details page containing a summary of properties and links to off-site repositories such as PubMed, GenBank, Entrez, and OMIM. The page provides item-specific information on position, cytoband, strand, data source, and encoded protein, mRNA, genomic sequence and alignment, as appropriate to the nature of the track.
A blue navigation bar at the top of the browser provides links to several other tools and data sources. For instance, under the "View" menu, the "DNA" link enables the user to view the raw genomic DNA sequence for the coordinate range displayed in the browser window. This DNA can encode track features via elaborate text formatting options. Other links tie the Genome Browser to the BLAT alignment tool, provide access to the underlying relational database via the Table Browser, convert coordinates across different assembly dates, and open the window at the complementary Ensembl or NCBI Genome Data Viewer annotation.
The browser data represents an immense collaborative effort involving thousands of people from the international biomedical research community. The UCSC Bioinformatics Group itself does no sequencing. Although it creates the majority of the annotation tracks in-house, the annotations are based on publicly available data contributed by many labs and research groups throughout the world. Several of the Genome Browser annotations are generated in collaboration with outside individuals or are contributed wholly by external research groups. UCSC's other major roles include building genome assemblies, creating the Genome Browser work environment, and serving it online. The majority of the sequence data, annotation tracks, and even software are in the public domain and are available for anyone to download.
In addition to the Genome Browser, the UCSC Genome Bioinformatics group provides several other tools for viewing and interpreting genome data:
- BLAT - a fast sequence-alignment tool similar to BLAST. Read more.
- Table Browser - convenient text-based access to the database underlying the Genome Browser. Read more.
- Genome Graphs - a tool that allows you to upload and display genome-wide data sets such as the results of genome-wide SNP association studies, linkage studies and homozygosity mapping. Read more.
- Gene Sorter - expression, homology, and other information on groups of genes that can be related in many ways. Read more.
Getting started: Genome Browser gateway
The UCSC Genome Bioinformatics home page provides access to Genome Browsers on several different genome assemblies. To get started, click the Browser link on the blue sidebar. This will take you to a Gateway page where you can select which genome to display. Note that there are also official mirror sites in Europe and Asia for users who are geographically closer to those continents than to the western United States.
Opening the Genome Browser at a specific position
To get oriented in using the Genome Browser, try viewing a gene or region of the genome with which you are already familiar, or use the default position. To open the Genome Browser window:
- Select the clade, genome and assembly that you wish to display from the corresponding pull-down menus. Assemblies are typically named by the first three characters of an organism's genus and species names. For older assemblies that are no longer available from the menu, the data may still be available on our Downloads page.
- Specify the genome location you'd like the Genome Browser to open to. To select a location, enter a valid position query in the search term text box at the top of the Gateway page or accept the default position already displayed. The search supports several different types of queries: gene symbols, mRNA or EST accession numbers, chromosome bands, descriptive terms likely to occur in GenBank text, or specific chromosomal ranges.
- Click the submit button to open up the Genome Browser window to the requested location. In cases where a specific term (accession, gene name, etc.) was queried, the item will be highlighted in the display.
Occasionally the Gateway page returns a list of several matches in response to a search, rather than immediately displaying the Genome Browser window. When this occurs, click on the item in which you're interested and the Genome Browser will open to that location.
The search mechanism is not a site-wide search engine. Instead, it primarily searches GenBank mRNA records whose text annotations can include gene names, gene symbols, journal title words, author names, and RefSeq mRNAs. Searches on other selected identifiers, such as NP and NM accession numbers, OMIM identifiers, and Entrez Gene IDs are supported. However, some types of queries will return an error, e.g. post-assembly GenBank entries, withdrawn gene names, and abandoned synonyms. If your initial query is unsuccessful, try entering a different related term that may produce the same location. For example, if a query on a gene symbol produces no results, try entering an mRNA accession, gene ID number, or descriptive words associated with the gene.
Finding a genome location using BLAT
If you have genomic, mRNA, or protein sequence, but don't know the name or the location to which it maps in the genome, the BLAT tool will rapidly locate the position by homology alignment, provided that the region has been sequenced. This search will find close members of the gene family, as well as assembly duplication artifacts. An entire set of query sequences can be looked up simultaneously when provided in fasta format.
A successful BLAT search returns a list of one or more genome locations that match the input sequence. To view one of the alignments in the Genome Browser, click the browser link for the match. The details link can be used to preview the alignment to determine if it is of sufficient match quality to merit viewing in the Genome Browser. If too many BLAT hits occur, try narrowing the search by filtering the sequence in slow mode with RepeatMasker, then rerunning the BLAT search.
For more information on conducting and fine-tuning BLAT searches, refer to the BLAT section of this document.
Opening the Genome Browser with a custom annotation track
You can open the Genome Browser window with a custom annotation track displayed by using the Add Custom Tracks feature available from the gateway and annotation tracks pages. For more information on creating and using custom annotation tracks, refer to the Creating custom annotation tracks section.
Annotation track data can be entered in one of three ways:
- Enter the file name for an annotation track source file in the Annotation File text box.
- Type or paste the annotation track data into the large text box.
- If the annotation data are accessible through a URL, enter the URL name in the large text box.
Once you've entered the annotation information, click the submit button at the top of the Gateway page to open up the Genome Browser with the annotation track displayed.
The Genome Browser also provides a collection of custom annotation tracks contributed by the UCSC Genome Bioinformatics group and the research community.
NOTE: If an annotation track does not display correctly when you attempt to upload it, you may need to reset the Genome Browser to its default settings, then reload the track. For information on troubleshooting display problems with custom annotation tracks, refer to the troubleshooting section in the Creating custom annotation tracks section.
Viewing genome data as text
The Table Browser, a portal to the underlying open source MariaDB relational database driving the Genome Browser, displays genomic data as columns of text rather than as graphical tracks. For more information on using the Table Browser, see the section Getting started: on the Table Browser.
Opening the Genome Browser from external gateways
Several external gateways provide direct links into the Genome Browser. Examples include: Entrez Gene, AceView, Ensembl, SuperFamily, and GeneCards. Journal articles can also link to the browser and provide custom tracks. Be sure to use the assembly date appropriate to the provided coordinates when using data from a journal source.
Tips for Use
To facilitate your return to regions of interest within the Genome Browser, save the coordinate range or bookmark the page of displays that you plan to revisit or wish to share with others.
It is usually best to work with the most recent assembly even though a full set of tracks might not yet be ready. Be aware that the coordinates of a given feature on an unfinished chromosome may change from one assembly to the next as gaps are filled, artifactual duplications are reduced, and strand orientations are corrected. The Genome Browser offers multiple tools that can correctly convert coordinates between different assembly releases. For more information on conversion tools, see the section Converting data between assemblies.
To ensure uninterrupted browser services for your research during UCSC server maintenance and power outages, bookmark a mirror site that replicates the UCSC genome browser.
Bear in mind that the Genome Browser cannot outperform the underlying quality of the draft genome. Assembly errors and sequence gaps may still occur well into the sequencing process due to regions that are intrinsically difficult to sequence. Artifactual duplications arise as unavoidable compromises during a build, causing misleading matches in genome coordinates found by alignment.
Interpreting and fine-tuning the Genome Browser display
The Genome Browser annotation tracks page displays a genome location specified through a Gateway search, a BLAT search, or an uploaded custom annotation track. There are five main features on this page: a set of navigation controls, a chromosome ideogram, the annotations tracks image, display configuration buttons, and a set of track display controls.
The first time you open the Genome Browser, it will use the application default values to configure the annotation tracks display. By manipulating the navigation, configuration and display controls, you can customize the annotation tracks display to suit your needs. For a complete description of the annotation tracks available in all assembly versions supported by the Genome Browser, see the Annotation Track Descriptions section.
The Genome Browser retains user preferences from session to session within the same web browser, although it never monitors or records user activities or submitted data. To restore the default settings, click the "Reset All User Settings" under the top blue Genome Browser menu. To return the display to the default set of tracks (but retain custom tracks and other configured Genome Browser settings), click the default tracks button on the Genome Browser page.
Annotation track display conventions
Annotation track descriptions: Each annotation track has an associated description page that contains a discussion of the track, the methods used to create the annotation, the data sources and credits for the track, and (in some cases) filter and configuration options to fine-tune the information displayed in the track. To view the description page, click on the mini-button to the left of a displayed track or on the label for the track in the Track Controls section.
Annotation track details pages: When an annotation track is displayed in full, pack, or squish mode, each line item within the track has an associated details page that can be displayed by clicking on the item or its label. The information contained in the details page varies by annotation track, but may include basic position information about the item, related links to outside sites and databases, links to genomic alignments, or links to corresponding mRNA, genomic, and protein sequences.
Gene prediction tracks: Coding exons are represented by blocks connected by horizontal lines representing introns. The 5' and 3' untranslated regions (UTRs) are displayed as thinner blocks on the leading and trailing ends of the aligning regions. In full display mode, arrowheads on the connecting intron lines indicate the direction of transcription. In situations where no intron is visible (e.g. single-exon genes, extremely zoomed-in displays), the arrowheads are displayed on the exon block itself.
Pattern Space Layout (PSL) alignment tracks: Aligning regions (usually exons when the query is cDNA) are shown as black blocks. In dense display mode, the degree of darkness corresponds to the number of features aligning to the region or the degree of quality of the match. In pack or full display mode, the aligning regions are connected by lines representing gaps in the alignment (typically spliced-out introns), with arrowheads indicating the orientation of the alignment, pointing right if the query sequence was aligned to the forward strand of the genome and left if aligned to the reverse strand. Two parallel lines are drawn over double-sided alignment gaps, which skip over unalignable sequence in both target and query. For alignments of ESTs, the arrows may be reversed to show the apparent direction of transcription deduced from splice junction sequences. In situations where no gap lines are visible, the arrowheads are displayed on the block itself. To prevent display problems, the Genome Browser imposes an upper limit on the number of alignments that can be viewed simultaneously within the tracks image. When this limit is exceeded, the Browser displays the best several hundred alignments in a condensed display mode, then lists the number of undisplayed alignments in the last row of the track. In this situation, try zooming in to display more entries or to return the track to full display mode. For some PSL tracks, extra coloring to indicate mismatching bases and query-only gaps may be available.
Chain tracks (2-species alignment): Chain tracks display boxes joined together by either single or double lines. The boxes represent aligning regions. Single lines indicate gaps that are largely due to a deletion in the genome of the first species or an insertion in the genome of the second species. Double lines represent more complex gaps that involve substantial sequence in both species. This may result from inversions, overlapping deletions, an abundance of local mutation, or an unsequenced gap in one species. In cases where there are multiple chains over a particular portion of the genome, chains with single-lined gaps are often due to processed pseudogenes, while chains with double-lined gaps are more often due to paralogs and unprocessed pseudogenes. In the fuller display modes, the individual feature names indicate the chromosome, strand, and location (in thousands) of the match for each matching alignment.
Net tracks (2-species alignment): Boxes represent ungapped alignments, while lines represent gaps. Clicking on a box displays detailed information about the chain as a whole, while clicking on a line shows information on the gap. The detailed information is useful in determining the cause of the gap or, for lower level chains, the genomic rearrangement. Individual items in the display are categorized as one of four types (other than gap):
- Top - The best, longest match. Displayed on level 1.
- Syn - Lineups on the same chromosome as the gap in the level above it.
- Inv - A lineup on the same chromosome as the gap above it, but in the opposite orientation.
- NonSyn - A match to a chromosome different from the gap in the level above.
Snake tracks: The snake alignment track (or snake track) shows the relationship between the chosen Browser genome (reference genome) and another genome (query genome). A snake is a way of viewing a set of pairwise gapless alignments that may overlap on both the reference and query genomes. Alignments are always represented as being on the positive strand of the reference species, but can be on either strand on the query sequence.
In full display mode, a snake track can be decomposed into two drawing elements: segments (colored rectangles) and adjacencies (lines connecting the segments). Segments represent subsequences of the target genome aligned to the given portion of the reference genome. Adjacencies represent the covalent bonds between the aligned subsequences of the target genome.
Red tick-marks within segments represent substitutions with respect to the reference, shown in windows of the reference of (by default) up to 50 Kb. Zoomed in to the base level, these substitutions are labeled with the non-reference base.
An insertion in the reference relative to the query creates a gap between abutting segment sides that is connected by an adjacency. An insertion in the query relative to the reference is represented by an orange tick-mark that splits a segment at the location the extra bases would be inserted. Simultaneous independent insertions in both query and reference look like an insertion in the reference relative to the target, except that the corresponding adjacency connecting the two segments is colored orange. More complex structural rearrangements create adjacencies that connect the sides of non-abutting segments in a natural fashion.
Pack mode can be used to display a larger number of snake tracks in the limited vertical browser. This mode eliminates the adjacencies from the display and forces the segments onto as few rows as possible, given the constraint of still showing duplications in the query sequence.
Dense mode further eliminates these duplications so that each snake track is compactly represented along just one row.
Wiggle tracks: These tracks plot a continuous function along a chromosome. Data is displayed in windows of a set number of base pairs in width. The score for each window displays as "mountain ranges" The display characteristics vary among the tracks in this group. See the individual track descriptions for more information on interpreting the display. If the peak is taller or shorter than what can be shown in the display, it is clipped and colored magenta.
Annotation track display modes
Each annotation track within the window may have up to five display modes:
- Hide: the track is not displayed at all. To hide all the annotation tracks, click the hide all button. This mode is useful for restricting the display to only those tracks in which you are interested. For example, someone who is not interested in SNPs or mouse synteny may want to hide these tracks to reduce track clutter and improve speed. There are a few annotation tracks that pertain only to one specific chromosome, e.g. Sanger22, Rosetta. In these cases, the track and its associated controller will be hidden automatically when the track window is not open to the relevant chromosome.
-
Dense: the track is displayed with all features collapsed into a single line.
This mode is useful for reducing the amount of space used by a track when you don't need
individual line item details or when you just want to get an overall view of an annotation. For
example, by opening an entire chromosome and setting the RefSeq Genes track to dense, you can get
a feel for the known gene density of the chromosome without displaying excessive detail.

-
Full: the track is displayed with each annotation feature on a separate line. It
is recommended that you use this option sparingly, due to the large number of individual track
items that may potentially align at the selected position. For example, hundreds of ESTs might
align with a specified gene. When the number of lines within a requested track location exceeds
250, the track automatically defaults to a more tightly-packed display mode. In this situation,
you can restore the track display to full mode by narrowing the chromosomal range displayed or by
using a track filter to reduce the number of items displayed. On tracks that contain only hide,
dense, and full modes, you can toggle between full and dense display modes by clicking on the
track's center label.

-
Squish: the track is displayed with each annotation feature shown separately, but
at 50% the height of full mode. Features are unlabeled, and more than one may be drawn on the same
line. This mode is useful for reducing the amount of space used by a track when you want to view a
large number of individual features and get an overall view of an annotation. It is particularly
good for displaying tracks in which a large number of features align to a particular section of a
chromosome, e.g. EST tracks.
-
Pack: the track is displayed with each annotation feature shown separately and
labeled, but not necessarily displayed on a separate line. This mode is useful for reducing the
amount of space used by a track when you want to view the large number of individual features
allowed by squish mode, but need the labeling and display size provided by full mode. When the
number of lines within the requested track location exceeds 250, the track automatically defaults
to squish display mode. In this situation, you can restore the track display to pack mode by
narrowing the chromosomal range displayed or by using a track filter to reduce the number of items
displayed. To toggle between pack and full display modes, click on the track's center label.

The track display controls are grouped into categories that reflect the type of data in the track, e.g., Gene Prediction Tracks, mRNA and EST tracks, etc. To change the display mode for a track, find the track's controller in the Track Controls section at the bottom of the Genome Browser page, select the desired mode from the control's display menu, and then click the refresh button. Alternatively, you can change the display mode by using the Genome Browser's right-click navigation feature, or can toggle between dense and full modes for a displayed track (or pack mode when available) by clicking on the optional center label for the track.
Changing the display mode for a group of tracks
Track display modes may be set individually or as a group on the Genome Browser Track Configuration page. To access the configuration page, click the configure button on the annotation tracks page or the configure tracks and display button on the Gateway page. Exercise caution when using the show all buttons on track groups or assemblies that contain a large number tracks; this may seriously impact the display performance of the Genome Browser or cause your Internet browser to time out.
Hiding the track display controls
The entire set of track display controls at the bottom of the annotation tracks page may be hidden from view by checking the Show track controls under main graphic option in the Configure Image section of the Track Configuration page.
Changing the display of a track by using filters and configuration options
Some tracks have additional filter and configuration capabilities, e.g., EST tracks, mRNA tracks, NC160, etc. These options let the user modify the color or restrict the data displayed within an annotation track. Filters are useful for focusing attention on items relevant to the current task in tracks that contain large amounts of data. For example, to highlight ESTs expressed in the liver, set the EST track filter to display items in a different color when the associated tissue keyword is "liver" Configuration options let the user adjust the display to best show the data of interest. For example, the min vertical viewing range value on wiggle tracks can be used to establish a data threshold. By setting the min value to "50", only data values greater than 50 percent will display.
To access filter and configuration options for a specific annotation track, open the track's description page by clicking the label for the track's control menu under the Track Controls section, the mini-button to the left of the displayed track, or the "Configure..." option from the Genome Browser's right-click popup menu. The filter and configration section is located at the top of the description page. In most instances, more information about the configuration options is available within the description text or through a special help link located in the configuration section.
Filter and configuration settings are persistent from session to session on the same web browser. To return the Genome Browser display to the default set of tracks (but retain custom tracks and other configured Genome Browser settings), click the default tracks button on the Genome Browser tracks page. To remove all user configuration settings and custom tracks, and completely restore the defaults, click the "Reset All User Settings" under the top blue Genome Browser menu.
Video tutorial on changing track display modes
Zooming and scrolling the tracks display
At times you may want to adjust the amount of flanking region displayed in the annotation tracks window or adjust the scale of the display. At a scale of 1 pixel per base pair, the window accurately displays the width of exons and introns, and indicates the direction of transcription (using arrowheads) for multi-exon features. At a grosser scale, certain features - such as thin exons - may disappear. Also, some exons may falsely appear to fall within RepeatMasker features at some scales.
Click the zoom in and zoom out buttons at the top of the Genome Browser page to zoom in or out on the center of the annotation tracks window by 1.5, 3 or 10-fold. Alternatively, you can zoom in 3-fold on the display by clicking anywhere on the Base Position track. In this case, the zoom is centered on the coordinate of the mouse click. To view the base composition of the sequence underlying the current annotation track display, click the base button.
Quickly zoom to a specific region of interest by using the browser's "drag-and-select" feature. To define the region you wish to zoom to, click and hold the mouse button on one edge of the desired zoom area in the Base Position track, drag the mouse right or left to highlight the selection area, then release the mouse button. A "drag-and-select" popup will appear. Click on the "Zoom In" button to zoom in on the selected region. To disable the drag-and-select popup, check the "Don't show this dialog again and always zoom" checkbox. To drag-and-select (zoom) on a part of the image other than the Base Position track, depress the shift key before clicking and dragging the mouse. Note that the Enable advanced javascript features option on the Track Configuration page must be toggled on to use this feature.
To scroll (pan) the view of the entire tracks image horizontally, click on the image and drag the cursor to the left or right, then release the mouse button, to shift the displayed region in the corresponding direction. The view may be scrolled by up to one image width. To scroll the annotation tracks horizontally by set increments of 10%, 50%, or 95% of the displayed size (as given in base pairs), click the corresponding move arrow. It is also possible to scroll the left or right side of the tracks by a specified number of vertical gridlines while keeping the position of the opposite side fixed. To do this, click the appropriate move start or move end arrow, located under the annotation tracks window. For example, to keep the left-hand display coordinate fixed but increase the right-hand coordinate, you would click the right-hand move end arrow. To increase or decrease the gridline scroll interval, edit the value in the move start or move end text box.
Highlighting a region
The browser's "drag-and-select" pop-up menu provides options to add single or multiple vertical highlights to selected regions, as described below:
Main features in drag-and-select menu:
- Use shift+drag or click-drag to enable the "drag-and-select" dialog box menu.
- In the menu, a checkbox controls behavior for drag-and-select; you can hide the menu and always zoom with shift+select. If selected, re-enable via 'View - Configure Browser' (keyboard shortcut: c then f).
- A "color picker" option allows for easy color selection of each highlight; you can also create multiple highlights (each with various colors if desired).
- Hold Shift+drag to show the menu (or click+drag).
- Hold Alt+drag to add a highlight (without displaying the menu).
- Hold Ctrl+drag (Windows) or Cmd+drag (Mac) to zoom (without displaying the menu).
- To cancel, press Esc anytime or drag mouse outside image.
- Highlight the current position with the keyboard shortcut "h then m."
- Clear all highlights with View - Clear Highlights (or keyboard shortcut h then c), or simply right-click on a highlight to remove all highlights.
In the genome browser, there are also options for right-clicking:
- Remove highlighting
- Zoom in to a highlighted region
- Highlight a gene - right-click on the gene (e.g., SOD1) and select "Highlight SOD1"
Changing the displayed track position
To display a completely different position in the genome, enter the new query in the position/search text box, then click the jump button. For more information on valid entries for this text box, refer to the Getting started section.
If a chromosome image (ideogram) is available above the track display, click anywhere on the chromosome to move to that position (the current window size will be maintained). Select a region of any size by clicking and dragging in the image. Finally, hold the "control" key while clicking on a chromosome band to select the entire band.
Changing the order of the displayed tracks
To vertically reposition a track in the annotation track window, click-and-hold the mouse button on the side label, then drag the highlighted track up or down within the image. Release the mouse button when the track is in the desired position. To move an entire group of associated tracks (such as all the displayed subtracks in a composite track), click-and-hold the gray mini-button to the left of the tracks, then drag.
Viewing multiple regions
To remove intronic or intergenic regions from the display or to view only custom specified regions, click the multi-region button under the track image. For human assemblies hg17 and later, you may also replace a section of the reference genome with an alternate haplotype chromosome in order to view annotations upstream and downstream of the sequence. For more information about the multi-region feature see the multi-region help page.
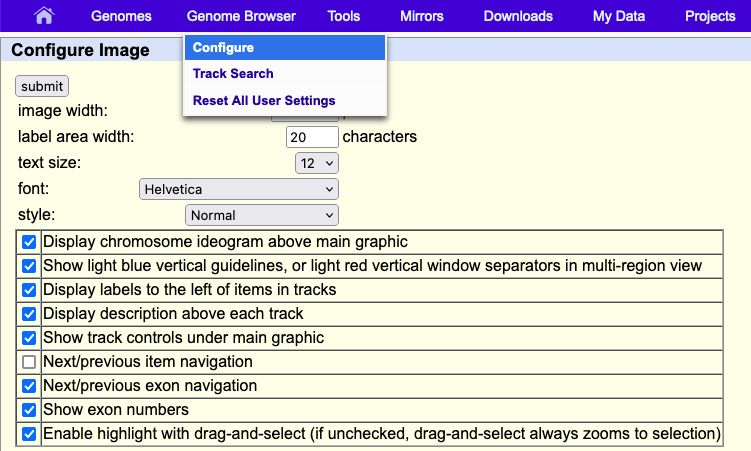
Configuration panel
Changing the width of the annotation track window
The first time the annotation track window is displayed, or after the Genome Browser has been reset, the size of the track window is set by default to the width that best fits your Internet browser window. If you horizontally resize the browser window, you can automatically adjust the annotation track image size to the new width by clicking the resize button under the track image. To manually override the default width, enter a new value in the image width text box on the Track Configuration page, then click the submit button. The maximum supported width is 5000 pixels.
Changing the width of the label area to the left of the image
The item labels (or track label, when viewed in dense mode) are displayed to the left of the annotation image. The width of this area is set to 17 characters by default. To change the width, edit the value in the label area width text box on the Track Configuration page, then click Submit.
Changing the text size, font, in the annotation track image
The annotation track image may be adjusted to display text in a range of fonts from AvantGarde, Courier, and Times. To change the size of the text, select an option from the text size pull-down menu on the Browser Configuration page, which you can find under the top blue "Genome Browser" menu by clicking Configure. Once you have made your selection, which can also include style click submit. The text size is set to "12" and "Helvetica" by default.
Hiding the chromosome ideogram
The chromosome ideogram, located just above the annotation tracks image, provides a graphical overview of the features on the selected chromosome, including its bands, the position of the centromere, and an indication of the region currently displayed in the annotation tracks image. To hide the ideogram, uncheck the Display chromosome ideogram above main graphic box on the Tracks Configuration page.
Hiding the light blue vertical guidelines
The light blue vertical guidelines on the annotation tracks image may be removed by unchecking the Show light blue vertical guidelines box on the Track Configuration page.
Hiding the annotation track labels and description
The track and element labels displayed above and to the left of the tracks in the annotation tracks image may be hidden from view by unchecking the Display track descriptions above each track and Display labels to the left of items in tracks boxes, respectively, on the Track Configuration page.
Enabling next/previous item and exon navigation
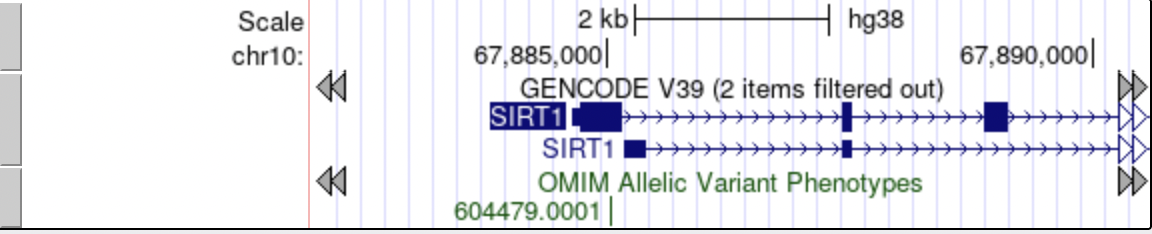
Gray arrows jump to the next item, while white arrows advance to the next exon.
When the Next/previous item navigation configuration option is toggled on, on the Track Configuration page, gray double-headed arrows display in the Genome Browser tracks image on both sides of the track labels of gene, mRNA and EST tracks (or any standard tracks based on BED, PSL or genePred format). Clicking on the gray arrows shifts the image window toward that end of the chromosome so that the next item in the track is displayed. Similarly, the Next/previous exon navigation configuration option displays white double-headed arrows on the end of any item that extends off the edge of the current image. Clicking on one of the white arrows shifts the image window to the next exon in the indicated direction, unless the image window interrupts an exon, in which case the window shifts to the edge of the current exon. If the image window happens to be within a 5' or 3' UTR, then clicking the arrows shifts the image window towards the start or end of the next coding region, not the end of the exon.
Using the right-click navigation feature
Several of the common display and navigation operations offered on the Genome Browser tracks page may be quickly accessed by right-clicking on a feature on the tracks image and selecting an option from the displayed popup menu.
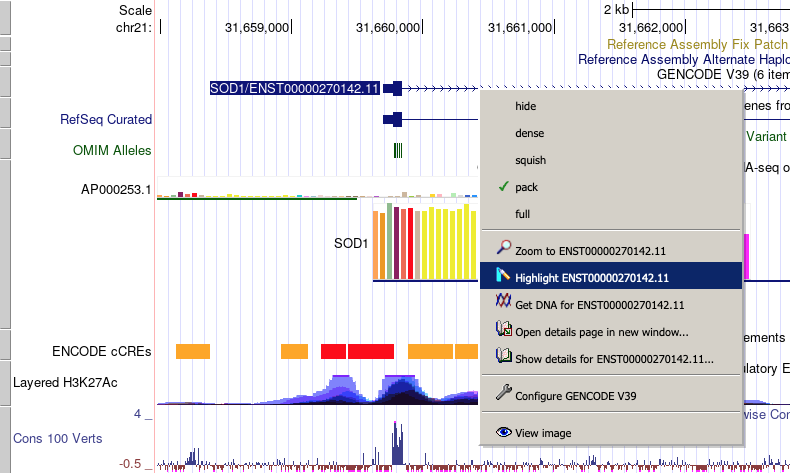
Depending on context, the right-click feature allows the user to:
- change the track display mode
- zoom in or out to the exact position coordinates of the feature
- highlight the feature
- open the "Get DNA for..." link for the feature's coordinates
- display details about the feature
- open a popup window to configure the track's display of the feature
- display the entire tracks image in a separate window for inclusion in spreadsheets or other documents. The Genome Browser PDF/PS option under the View menu can also be used to generate a high-quality annotation tracks image suitable for printing.
To use the right-click feature, make sure your internet browser allows the display of popup windows from genome.ucsc.edu. When enabled, the right-click navigation feature replaces the default contextual popup menu typically displayed by the Internet browser when a user right-clicks on the tracks image. A few combinations of the Mozilla Firefox browser on Mac OS do not support the right-click menu functionality using secondary click; in these instances, ctrl+left-click must be used to display the menu.
Printing a copy of the annotation track window
The Genome Browser provides a mechanism for saving a copy of the currently displayed annotation tracks image to a file that can be printed or edited. Images saved in PostScript format can be printed at high resolution and edited by drawing programs such as Adobe Illustrator. This is useful for generating figures intended for publication. Images can also be saved in PDF format for viewing by Adobe Acrobat Reader.
To print or save the image to a file:
- In the blue navigation bar at the top of the screen, from the "View" menu, click the "PDF/PS" link.
- Click one of the PDF or EPS links.
NOTE: If you have configured your browser image to use one of the larger font sizes, the text in the resulting screen shot may not display correctly. If you encounter this problem, reduce the Genome Browser font size using the Configuration utility, then repeat the save/print process.
Using BLAT alignments
BLAT (BLAST-Like Alignment Tool) is a very fast sequence alignment tool similar to BLAST. For more information on BLAT's internal scoring schemes and its overall n-mer alignment seed strategy, refer to W. James Kent (2002) BLAT - The BLAST-Like Alignment Tool, Genome Res 12:4 656-664.
On DNA queries, BLAT is designed to quickly find sequences with 95% or greater similarity of length 25 bases or more. It may miss genomic alignments that are more divergent or shorter than these minimums, although it will find perfect sequence matches of 32 bases and sometimes as few as 22 bases. The tool is capable of aligning sequences that contain large introns. On protein queries, BLAT rapidly locates genomic sequences with 80% or greater similarity of length 20 amino acids or more. In general, gene family members that arose within the last 350 million years can generally be detected. More divergent sequences can be aligned to the human genome by using NCBI's BLAST and psi-BLAST, then using BLAT to align the resulting match onto the UCSC genome assembly. In practice DNA Blat works well on primates, and protein Blat works well on land vertebrates.
Some common uses of BLAT include:
- finding the genomic coordinates of mRNA or protein within a given assembly
- determining the exon structure of a gene
- displaying a coding region within a full-length gene
- isolating an EST of special interest as its own track
- searching for gene family members
- finding human homologs of a query from another species
- finding homologs of a query in all species hosted on the UCSC Genome Browser
Making a BLAT query
To locate a nucleotide or protein within a genome using BLAT:
- Open the BLAT Search Genome page by clicking on the "Tools" pulldown in the top blue menu bar of the Genome Browser.
-
Blat a single genome – Select the genome, assembly, query type, output sort order, and
output type. To order the search
results based on the closeness of the sequence match, choose one of the score options in the
Sort output menu. The score is determined by the number of matches vs. mismatches in the
final alignment of the query to the genome.
Blat ALL genomes – The "Search ALL" checkbox above the Genome drop-down list allows you to search the genomes of the default assemblies for all of our organisms. This shows you an ordered list of the default assemblies having the greatest similarity with your query sequence. Additionally, the Search ALL feature searches any attached hubs' blat servers, meaning you can search your user-generated assembly hubs. - If the sequence to be uploaded is in an unformatted plain text file, enter the file name in the Upload sequence text box, then click the submit file button. Otherwise, paste the sequence or fasta-formatted list into the large edit box, and then click the submit button. Input sequence can be obtained from the Genome Browser as well as from a custom annotation track.
Header lines may be included in the input text if they are preceded by > and contain unique names. Multiple sequences may be submitted at the same time if they are of the same type and are preceded by unique header lines. Numbers, spaces, and extraneous characters are ignored:
>sequence_1
ATGCAGAGCAAGGTGCTGCTGGCCGTCGCCCTGTGGCTCTGCGTGGAGAC
CCGGGCCGCCTCTGTGGGTTTGCCTAGTGTTTCTCTTGATCTGCCCAGGC
>sequence_2
ATGTTGTTTACCGTAAGCTGTAGTAAAATGAGCTCGATTGTTGACAGAGA
TGACAGTAGTATTTTTGATGGGTTGGTGGAAGAAGATGACAAGGACAAAG
>sequence_3
ATGCTGCGAACAGAGAGCTGCCGCCCCAGGTCGCCCGCCGGACAGGTGGC
CGCGGCGTCCCCGCTCCTGCTGCTGCTGCTGCTGCTCGCCTGGTGCGCGG
BLAT limitations
DNA input sequences are limited to a maximum length of 25,000 bases. Protein or translated input sequences must not exceed 10,000 letters. As many as 25 multiple sequences may be submitted at the same time. The maximum combined length of DNA input for multiple sequence submissions is 50,000 bases (with a 25,000 base limit per individual sequence). For protein or translated input, the maximum combined input length is 25,000 letters (with a 5000 letter limit per individual sequence).
NOTE: Program-driven BLAT use is limited to a maximum of one hit every 15 seconds and no more than 5000 hits per day.
BLAT query search results
If a query returns successfully, BLAT will display a flat database file that summarizes the alignments found. A BLAT query often generates multiple hits. This can happen when the genome contains multiple copies of a sequence, paralogs, pseudogenes, statistical coincidences, artifactual assembly duplications, or when the query itself contains repeats or common retrotransposons. When too many hits occur, try resubmitting the query sequence after filtering in slow mode with RepeatMasker.
Items in the search results list are ordered by the criteria specified in the Sort output menu. Each line item provides links to view the details of the sequence alignment or to open the corresponding view in the Genome Browser. The details link gives the letter-by-letter alignment of the sequence to the genome. It is recommended that you first examine the details of the alignment for match quality before viewing the sequence in the Genome Browser.
When several nearby BLAT matches occur on a single chromosome, a simple trick can be used to quickly adjust the Genome Browser track window to display all of them: open the Genome Browser with the match that has the lowest chromosome start coordinate, paste in the highest chromosome end coordinate from the list of matches, then click the jump button.
Creating a custom annotation track from BLAT output
To make a custom track directly from BLAT, select the PSL format output option. The resulting PSL track can be uploaded into the Genome Browser by pasting the data into the data text box on the Genome Browser Add Custom Tracks page, accessed via the "add custom tracks" button on the Browser gateway and annotation tracks pages. See the Creating custom annotation tracks section for more information.
Using BLAT for large batch jobs or commercial use
For large batch jobs or internal parameter changes, it is best to install command line BLAT on your own Linux server. Sources and executables are free for academic, personal, and non-profit purposes. BLAT source may be downloaded from https://genome-test.gi.ucsc.edu/~kent/src/ (look for the blatSrc*.zip file with the most recent date). For BLAT executables, go to http://genome-test.soe.ucsc.edu/~kent/exe/; binaries are sorted by platform. Non-exclusive commercial licenses are available from the Kent Informatics website.
BLAT documentation
For more information on the BLAT suite of programs, see the BLAT Program Specifications and the Blat section of the Genome Browser FAQ.
Annotation track descriptions
Detailed information about an individual annotation track, including display characteristics, configuration information, and associated database tables, may be obtained from the track description page accessed by clicking the mini-button to the left of the displayed track in the Genome Browser, or by selecting the "Open details..." or "Show details..." option from the Genome Browser's right-click menu. Click the "View table schema" link on the track description page to display additional information about the primary database table underlying the track. Table schema information may also be accessed via the "describe table schema" button in the Table Browser. For more information on configuring and using the tracks displayed in the Genome Browser track window, see the section Interpreting and Fine-tuning the Genome Browser display.
Tips for viewing annotation track data
- To display a description page with more information about the track, click on the mini-button to the left of a track.
- To display a details page with additional information about a specific line item within a track in full display mode, click on the item or its label.
- A track does not appear in the browser if its display mode is set to hide. To restrict the browser's display to only those tracks in which you're interested, set the display mode of the unwanted tracks to hide.
- A track set to full display mode will default to a more tightly packed display mode if the total number of lines in the track exceeds 250.
- To quickly toggle between full and dense or pack display modes, click on the track's center label.
- Only the most recent assemblies are fully active. The data for older assemblies may be available on our Downloads page.
- Not all tracks appear in all assemblies. Only a basic set of tracks appears initially in a new assembly.
- Track data can be viewed as text tables using the Table Browser.
- Credit goes to many individuals and institutions for generously contributing the tracks. For specific information about the contributors of a given track, look at the Credits section on a track's description page.
Getting started on the Table Browser
The Table Browser provides text-based access to the genome assemblies and annotation data stored in the Genome Browser database. As a flexible alternative to the graphical-based Genome Browser, this tool offers an enhanced level of query support that includes restrictions based on field values, free-form SQL queries, and combined queries on multiple tables. Output can be filtered to restrict the fields and lines returned, and may be organized into one of several formats, including a simple tab-delimited file that can be loaded into a spreadsheet or database as well as advanced formats that may be uploaded into the Genome Browser as custom annotation tracks. The Table Browser provides a convenient alternative to downloading and manipulating the entire genome and its massive data tracks. (See the Downloading Genome Data section.)
For information on using the Table Browser features, refer to the Table Browser User Guide.
Getting started using Sessions
The Sessions tool allows users to configure their browsers with specific track combinations, including custom tracks, and save the configuration options. Multiple sessions may be saved for future reference, for comparison of scenarios or for sharing with colleagues. Saved sessions will not be expired, however we still recommend that you keep local back-ups of your session contents and any associated custom tracks. User-generated tracks can be saved within sessions.
This tool may be accessed by clicking the "My Data" pulldown in the top blue navigation bar in any assembly and then selecting Sessions. To ensure privacy and security, you must create an account and/or log in to use the Session tool. Individual sessions may be designated by the user as either "shared" or "non-shared" to protect the privacy of confidential data. To avoid having a new shared session from someone else override existing Genome Browser settings, users are encouraged to open a new web-browser instance or to save existing settings in a session before loading a new shared session.
For more detailed information on using the Session tool, see the Sessions User Guide.
Getting started on Genome Graphs
The Genome Graphs tool can be used to display genome-wide data sets such as the results of genome-wide SNP association studies, linkage studies, and homozygosity mapping. This tool is not pre-loaded with any sample data; instead, you can upload your own data for display by the tool.
Once you have uploaded your data, you can view it in a variety of ways. You can view multiple sets of genome-wide data simultaneously either as superimposed graphs or side-by-side graphs. Once you see an area of interest in the Genome Graphs view, you can click on it to go directly to the Genome Browser at that position. You can also set a significance threshold for your data and view only regions or gene sets that meet that threshold.
For information on using the Genome Graphs features, refer to the Genome Graphs User Guide.
Using the VisiGene Image Browser
VisiGene is a browser for viewing in situ images. It enables the user to examine cell-by-cell as well as tissue-by-tissue expression patterns. The browser serves as a virtual microscope, allowing users to retrieve images that meet specific search criteria, then interactively zoom and scroll across the collection.
To start the VisiGene browser, click the VisiGene link in the left-hand sidebar menu on the Genome Browser home page.
Images Available
The following image collections are currently available for browsing:
- High-quality high-resolution images of eight-week-old male mouse sagittal brain slices with reverse-complemented mRNA hybridization probes from the Allen Brain Atlas, courtesy of the Allen Institute for Brain Science
- Mouse in situ images from the Jackson Lab Gene Expression Database (GXD) at MGI
- Transcription factors in mouse embryos from the Mahoney Center for Neuro-Oncology
- Mouse head and brain in situ images from NCBI's Gene Expression Nervous System Atlas (GENSAT) database
- Xenopus laevis in situ images from the National Institute for Basic Biology (NIBB) XDB project
Searching the Image Database
The image database may be searched by gene symbols, authors, years of publication, body parts, GenBank or UniProtKB accessions, organisms, Theiler stages (mice), and Nieuwkoop/Faber stages (frogs). The search returns only those images that match all the specified criteria. For a list of sample search strings, see the VisiGene Gateway page.
The wildcard characters * and ? are supported for gene name searches. For example, to view the images of all genes in the Hox A cluster, search for hoxa*. When searching on author names that include initials, use the format Smith AJ.
Image Navigation
Following a successful search, VisiGene displays a list of thumbnails of images matching the search criteria in the lefthand pane of the browser. By default, the image corresponding to the first thumbnail in the list is displayed in the main image pane. If more than 25 images meet the search criteria, links at the bottom of the thumbnail pane allow the user to toggle among pages of search results. To display a different image in the main browser pane, click the thumbnail of the image you wish to view.
By default, an image is displayed at a resolution that provides optimal viewing of the overall image. This size varies among images. The image may be zoomed in or out, sized to match the resolution of the original image or best fit the image display window, and moved or scrolled in any direction to focus on areas of interest. The original full-sized image may also be downloaded.
Zooming in: To enlarge the image by 2X, click the Zoom in button above the image or click on the image using the left mouse button. Alternatively, the + key may be used to zoom in when the main image pane is the active window.
Zooming out: To reduce the image by 2X, click the Zoom out button above the image or click on the image using the right mouse button. Alternatively, the - key may be used to zoom out when the main image pane is the active window.
Sizing to full resolution: Click the Zoom full button above the image to resize the image such that each pixel on the screen corresponds to a pixel in the digitized image.
Sizing to best fit: Click the Zoom fit button above the image to zoom the image to the size that best fits the main image pane.
Moving the image: To move the image viewing area in any direction, click and drag the image using the mouse. Alternatively, the following keyboard shortcuts may be used after clicking on the image:
- Scroll left in the image: Left-arrow key or Home key
- Scroll right in the image: Right-arrow key or End key
- Scroll up in the image: Up-arrow key or PgUp key
- Scroll down in the image: Down-arrow key or PgDn key
Downloading the original full-sized image: Most images may be viewed in their original full-sized format by clicking the "download" link at the bottom of the image caption. NOTE: due to the large size of some images, this action may take a long time and could potentially exceed the capabilities of some Internet browsers.
If you have an image set you would like to contribute for display in the VisiGene Browser, contact Jim Kent.
DNA text formatting
The Genome Browser provides a feature to configure the retrieval, formatting, and coloring of the text used to depict the DNA sequence underlying the features in the displayed annotation tracks window. Retrieval options allow the user to add a padding of extra bases to the upstream or downstream end of the sequence. Formatting options range from simply displaying exons in upper case to elaborately marking up a sequence according to multiple track data. The DNA sequence covered by various tracks can be highlighted by case, underlining, bold or italic fonts, and color.
The DNA display configuration feature can be useful to highlight features within a genomic sequence, point out overlaps between two types of features (for example, known genes vs. gene predictions), or mask out unwanted features.
Using the DNA text formatting feature
To access the feature, click on the "View" pulldown on the top blue menu bar on the Genome Browser page and select "DNA", or select the "Get DNA..." option from the Genome Browser's right-click menu depending on context. "The Get DNA in Window" page that appears contains sections for configuring the retrieval and output format.
To display extra bases upstream of the 5' end of your sequence or downstream of the 3' end of the sequence, enter the number of bases in the corresponding text box. This option is useful in looking for regulatory regions.
The Sequence Formatting section lists several options for adjusting the case of all or part of the DNA sequence. To choose one of these formats, click the corresponding option button, then click the get DNA button. To access a table of extended formatting options, click the Extended case/color options button.
The Extended DNA Case/Color page presents a table with many more format options. The page provides instructions for using the formatting table, as well as examples of its use. The list of tracks in the Track Name column is automatically generated from the list of tracks available on the current genome.
Tips for Use
A few caveats mentioned on the Extended DNA Case/Color page bear repeating. Keep the formatting simple at first: it is easy to make a display that is pretty to look at but is also completely cryptic. Also, be careful when requesting complex formatting for a large chromosomal region: when all the HTML tags have been added to the output page, the file size may exceed the size limits that your Internet browser, clipboard, and other software can safely display. The maximum size of genome that can be formatted by the tool is approximately 10 Mbp.
Converting data between assemblies
Coordinates of features frequently change from one assembly to the next as gaps are closed, strand orientations are corrected, and duplications are reduced. Occasionally, a chunk of sequence may be moved to an entirely different chromosome as the map is refined. There are three different methods available for migrating data from one assembly to another: BLAT alignment, coordinate conversion, and coordinate lifting. The BLAT alignment tool is described in the section Using BLAT alignments.
Coordinate conversion
The Genome Browser Convert utility is useful for locating the position of a feature of interest in a different release of the same genome or (in some cases) in a genome assembly of another species. During the conversion process, portions of the genome in the coordinate range of the original assembly are aligned to the new assembly while preserving their order and orientation. In general, it is easier to achieve successful conversions with shorter sequences.
When coordinate conversion is available for an assembly, click on the "View" pulldown on the top blue menu bar on the Genome Browser page and select the "In Other Genomes (Convert)" link. You will be presented with a list of the genome/assembly conversion options available for the current assembly. Select the genome and assembly to which you'd like to convert the coordinates, then click the button. If the conversion is successful, the browser will return a list of regions in the new assembly, along with the percent of bases and span covered by that region. Click on a region to display it in the browser. If the conversion is unsuccessful, the utility returns a failure message.
Lifting coordinates
The liftOver tool is useful if you wish to convert a large number of coordinate ranges between assemblies. This tool is available in both web-based and command line forms, and supports forward/reverse conversions as well as conversions between species. You can use the BED format (e.g. "chr4 100000 100001", 0-based) or directly paste text from the position box ("chr4:100,001-100,001", 1-based). See our Coordinate Counting blog post for a discussion of the difference. If the coordinates do not cover a single base pair e.g. "chr4 100000 100000" (BED) or "chr4:100,001-100,000" (text), this tool automatically extends them to at least one base pair.
Note: It is not recommeneded to use LiftOver to convert SNPs between assemblies, and more information about how to convert SNPs between assemblies can be found on the following FAQ entry.
Web-based coordinate lifting
To access the graphical version of the liftOver tool, click on "Tools" pulldown in the top blue menu bar of the Genome Browser, then select LiftOver from the menu. Note that the web tool has an input file size limit of 500Mb, larger files will require using the command-line version.
To convert one or more coordinate ranges using the default conversion settings:
- Select the genome and assembly from which the ranges were taken ("Original"), as well as the genome and assembly to which the coordinates should be converted ("New").
- Select the Data Format option: Browser Extensible Data format (BED) or position (coordinates of the form chrN:start-end).
- Enter coordinate ranges in the selected data format into the large text box, one per line.
- Click Submit.
Alternatively, you may load the coordinate ranges from an existing data file by entering the file name in the upload box at the bottom of the screen, then clicking the Submit File button.
The default parameter settings are recommended for general purpose use of the liftOver tool. However, you may want to customize settings if you have several very large regions to convert.
Command-line coordinate lifting
The command-line version of liftOver offers the increased flexibility and performance gained by running the tool on your local server. See an example of running the liftOver tool on the command line. This utility requires access to a Linux platform. The executable file may be downloaded here. Command-line liftOver requires a UCSC-generated over.chain file as input. Pre-generated files for a given assembly can be accessed from the assembly's "LiftOver files" link on the Downloads page. If the desired conversion file is not listed, send a request to the genome mailing list and we may be able to generate one for you. For use of the command-line version of LiftOver, we require all for-profit businesses or commercial companies to purchase a license to support our small team.
Downloading genome data
Most of the underlying tables containing the genomic sequence and annotation data displayed in the Genome Browser can be downloaded. All of the tables are freely usable for any purpose except as indicated in the README.txt file in the download directories. This data was contributed by many researchers, as listed on the Genome Browser Credits page. Please acknowledge the contributor(s) of the data you use.
Downloading the data
Genome data can be downloaded in different ways using our North American and European download servers, hgdownload, hgdownload2, and hgdownload-euro.
- Via rsync:
- The UCSC Genome Browser hgdownload server contains download directories for all genome versions currently accessible in the Genome Browser. Either of the following rsync commands can quickly and efficiently download large files to your current directory (./).
- To download an entire directory (note the trailing slash), you would use an expression such as:
- For more information please click here.
- Via ftp:
-
The UCSC Genome Browser FTP server contains download directories for all genome versions
currently accessible in the Genome Browser. The FTP URLs
ftp://hgdownload.soe.ucsc.edu/goldenPath/,ftp://hgdownload2.soe.ucsc.edu/goldenPath/, orftp://hgdownload-euro.soe.ucsc.edu/goldenPath/will take you to a directory that contains the genome download directories. This download method is not recommended if you plan to download a large file or multiple files from a single directory compared to rsync (see above). You can, however, use themgetcommand to download multiple files:mget filename1 filename2, ormget -a(to download all the files in the directory). - Via the Downloads link:
- Click the Downloads link on the left side bar on the UCSC Genome Browser home page to display a list of all database directories available for download. If the data you wish to download pre-dates the assembly versions listed, look for the data on our Downloads page.
- Via the REST API:
- The REST API can be used to query both annotation and sequence data from any UCSC genome assembly or hub.
rsync -a -P rsync://hgdownload.soe.ucsc.edu/path/file ./rsync -a -P rsync://hgdownload2.soe.ucsc.edu/path/file ./rsync -a -P rsync://hgdownload-euro.soe.ucsc.edu/path/file ./rsync -a -P rsync://hgdownload.soe.ucsc.edu/directory/ ./rsync -a -P rsync://hgdownload2.soe.ucsc.edu/directory/ ./rsync -a -P rsync://hgdownload-euro.soe.ucsc.edu/directory/ ./Types of data available
There may be several download directories associated with each version of a genome assembly: the full data set (bigZips), the full data set by chromosome (chromosome), the annotation database tables (database), and one or more sets of comparative cross-species alignments.
BigZips contains the entire draft of the genome in chromosome and/or contig form. Depending on the genome, this directory may contain some or all of the following files:
- chromAgp.zip: Description of how the assembly was generated, unpacking to one file per chromosome.
- chromFa.zip: The assembly sequence chromosomes, in one file per chromosome. Repeats from RepeatMasker and Tandem Repeats Finder are shown in lower case; non-repeating sequence is in upper case. The main assembly is contained in the chrN.fa files, where chrN is the name of the chromosome. The chrN_random.fa files contain clones that are not yet finished or cannot be placed with certainty at a specific place on the chromosome. In some cases, including the human HLA region on chromosome 6, the chrN_random.fa files also contain haplotypes that differ from the main assembly.
- chromFaMasked.zip: The assembly sequence chromosomes, in one file per chromosome. Repeats are masked by capital Ns; non-repeating sequence is shown in upper case.
- chromOut.zip: RepeatMasker .out file for chromosomes, generated by RepeatMasker at the -s sensitive setting.
- chromTrf.zip: Tandem Repeats Finder locations, filtered to keep repeats with period less than or equal to 12, translated into one .bed file per chromosome.
- contigAgp.zip: Description of how the assembly was generated from fragments at a contig layout level.
- contigFa.zip: The assembly sequence contigs, in one file per contig. All contigs are in forward orientation relative to the chromosome. In some cases, this means that contigs will be reversed relative to their orientation in the NCBI assembly. Repeats are shown in lower case; non-repeating sequence is shown in upper case.
- contigFaMasked.zip: The assembly sequence contigs, in one file per contig. Repeats are masked by capital Ns; non-repeating sequence is shown in upper case.
- contigOut.zip: RepeatMasker .out file for contigs, generated by RepeatMasker at the -s sensitive setting.
- contigTrf.zip: Tandem Repeats Finder locations, filtered to keep repeats with period less than or equal to 12, and translated into one .bed file per contig.
- database.zip: The Genome Browser database as tab-delimited files and associated MariaDB (MySQL) table-creation tiles (eliminated in later assemblies due to size restrictions).
- est.fa.zip: Sequences of all GenBank ESTs for the selected species.
- liftAll.zip: The offsets of contigs within chromosomes.
- mrna.zip: mRNAs in GenBank from the selected species.
- refmrna.zip: RefSeq mRNAs from the selected species.
- upstream1000.zip: Sequences 1000 bases upstream of annotated transcription start of RefSeq genes. This includes only cases where the transcription start is annotated separately from the coding region start.
- upstream2000.zip: Same as upstream1000, but with 2000 bases.
- upstream5000.zip: Same as upstream1000, but with 5000 bases.
- xenoMrna.zip: All GenBank mRNAs from species other than that of the selected one.
Chromosomes contains the assembled sequence for the genome in separate files for each chromosome in a zipped fasta format. The main assembly can be found in the chrN.fa files, where N is the name of the chromosome. The chrN_random.fa files contain clones that are not yet finished or cannot be placed with certainty at a specific place on the chromosome. In some cases, the chrN_random.fa files also contain haplotypes that differ from the main assembly.
Database contains all of the positional and non-positional tables in the genome annotation database. Each table is represented by 2 files:
- .sql file: the SQL commands used to create the table.
- .txt.gz file: the MariaDB database table data in tab-delimited format and compressed with gzip.
Schema descriptions for all tables in the genome annotation database may be viewed by using the "describe table schema" button in the Table Browser.
Cross-species alignments directories, such as the vsMm4 and humorMm3Rn3 directories in the hg16 assembly, contain pairwise and multiple species alignments and filtered alignment files used to produce cross-species annotations. For more information, refer to the READMEs in these directories and the description of the Multiple Alignment Format (MAF).
Creating custom annotation tracks
The Genome Browser provides dozens of aligned annotation tracks that have been computed at UCSC or have been provided by outside collaborators. In addition to these standard tracks, it is also possible for users to upload their own annotation data for temporary display in the browser. These custom annotation tracks are viewable only on the machine from which they were uploaded and are automatically discarded 48 hours after the last time they are accessed, unless they are saved in a Session. Optionally, users can make custom annotations viewable by others as well. For a more stable option for custom annotations, we suggest using track hubs. A third, more technical, option is to operate a mirror. Custom tracks work well for quickly displaying data, while track hubs are more configurable and permanent.
Custom tracks are a wonderful tool for research scientists using the Genome Browser. Because space is limited in the Genome Browser track window, many excellent genome-wide tracks cannot be included in the standard set of tracks packaged with the browser. Other tracks of interest may be excluded from distribution because the annotation track data is too specific to be of general interest or can't be shared until journal publication. In the past, many individuals and labs contributed custom tracks to the Genome Browser website for use by others. To view a list of these custom annotation tracks, click here.
Track hubs are now the preferred approach for viewing and sharing data on the Browser. Labs, consortia, and institutions submit their hubs to be listed as a Public Hub. Track hubs require remotely hosted data. They use binary index files which allow the browser to quickly access only what is relevant for the current region being viewed in the browser. See the track hub help page for more information.
Genome Browser annotation tracks are based on files in line-oriented format. Each line in the file defines a display characteristic for the track or defines a data item within the track. Annotation files contain three types of lines: browser lines, track lines, and data lines. Empty lines and those starting with "#" are ignored.
To construct an annotation file and display it in the Genome Browser, follow these steps:
Step 1. Format the data set:
Format your data as a tab-separated file using one of the formats supported by the Genome
Browser. Annotation data can be in standard
GFF format or in a format designed specifically for the Human Genome Project or UCSC Genome Browser, including
bedGraph,
GTF,
PSL,
BED,
bigBed,
WIG,
bigGenePred,
bigNarrowPeak,
bigMaf,
bigChain,
bigPsl,
barChart,
bigBarChart,
interact,
bigInteract,
bigWig,
BAM,
CRAM,
VCF,
MAF,
BED detail,
Personal Genome SNP,
broadPeak,
narrowPeak,
and microarray
(BED15). GFF and GTF files must be tab-delimited rather than space-delimited to display
correctly. Chromosome references must be of the form chrN (the parsing of chromosome names
is case-sensitive). You may include more than one data set in your
annotation file; these need not be in the same format.
Step 2. Define the Genome Browser display characteristics:
Add one or more optional browser lines to the beginning of your formatted data
file to configure the overall display of the Genome Browser when it initially shows your annotation
data. Browser lines allow you to configure such things as the genome position that the Genome
Browser will initially open to, the width of the display, and the configuration of the other
annotation tracks that are shown (or hidden) in the initial display. NOTE: If the browser position
is not explicitly set in the annotation file, the initial display will default to the position
setting most recently used by the user, which may not be an appropriate position for viewing the
annotation track.
Step 3. Define the annotation track display characteristics:
Following the browser lines--and immediately preceding the formatted data--add a
track line to define the display attributes for your annotation data set. Track
lines enable you to define annotation track characteristics such as the name, description, colors,
initial display mode, use score, etc. The track type=<track_type>
attribute is required for some tracks. If you have included more than one data set in your
annotation file, insert a track line at the beginning of each new set of data.
Example #1: Here is an example of a simple annotation file that contains a list of chromosome coordinates.
browser position chr22:20100000-20100900
track name=coords description="Chromosome coordinates list" visibility=2
#chrom chromStart chromEnd
chr22 20100000 20100100
chr22 20100011 20100200
chr22 20100215 20100400
chr22 20100350 20100500
chr22 20100700 20100800
chr22 20100700 20100900Click here to view this track in the Genome Browser.
Example #2: Here is an example of an annotation file that defines 2 separate annotation tracks in BED format. The first track displays blue one-base tick marks every 10000 bases on chr22. The second track displays red 100-base features alternating with blank space in the same region of chr22.
browser position chr22:20100000-20140000
track name=spacer description="Blue ticks every 10000 bases" color=0,0,255,
#chrom chromStart chromEnd
chr22 20100000 20100001
chr22 20110000 20110001
chr22 20120000 20120001
track name=even description="Red ticks every 100 bases, skip 100" color=255,0,0
#chrom chromStart chromEnd name
chr22 20100000 20100100 first
chr22 20100200 20100300 second
chr22 20100400 20100500 thirdClick here to view this track in the Genome Browser.
Example #3a: This example shows an annotation file containing one data set in BED format. The track displays features with multiple blocks, a thick end and thin end, and hatch marks indicating the direction of transcription. The track labels display in green (0,128,0), and the gray level of the each feature reflects the score value of that line.
browser position chr22:1000-10000
browser hide all
track name="BED track" description="BED format custom track example" visibility=2 color=0,128,0 useScore=1
#chrom chromStart chromEnd name score strand thickStart thickEnd itemRgb blockCount blockSizes blockStarts
chr22 1000 5000 itemA 960 + 1100 4700 0 2 1567,1488, 0,2512
chr22 2000 7000 itemB 200 - 2200 6950 0 4 433,100,550,1500 0,500,2000,3500 Click here to view this track in the Genome Browser.
Example #3b: This example shows a simple annotation file containing one data set in the bigBed format. This track displays random sized blocks across chr21 in the human genome. The big data formats, such as the bigBed format, can be uploaded using a bigDataUrl that is specified in the track line. For more information on these track line parameters, refer to the Track Lines section. When using bigDataUrls, data is cached and updated every 300 seconds. If you are updating your big data tracks with different displays and are not seeing your track changes in the browser, you may want to add the udcTimeout= parameter to prevent caching of your track data and force a reload.
You may paste these two lines directly into the "Add Custom Tracks" page to view this example in the browser:
browser position chr21:33,031,597-33,041,570
track type=bigBed name="bigBed Example One" description="A bigBed file" bigDataUrl=http://genome.ucsc.edu/goldenPath/help/examples/bigBedExample.bbAlternatively, you may also upload just the URL of the bigBed file:
http://genome.ucsc.edu/goldenPath/help/examples/bigBedExample.bb This will infer the track type as "bigBed" based on the file extension and set the track name to "bigBedExample".
Step 4. Display your annotation track in the Genome Browser
From the Gateway page, select the
genome assembly on which your annotation data is based, click 'GO', then click the "add custom
tracks" or "manage custom tracks" button below the tracks window.
On the Add Custom Tracks page, load the annotation track data or URL for your custom track into the upper text box and the track documentation (optional) into the lower text box, then click the "Submit" button. Tracks may be loaded by entering text, a URL, or a pathname on your local computer. The track type=<track_type> attribute is required for some tracks. For more information on these methods, as well as information on creating and adding track documentation, see Loading a Custom Track into the Genome Browser.
If you encounter difficulties displaying your annotation, read the section Troubleshooting Annotation Display Problems.
Step 5. (Optional) Add details pages for individual track features
After you've constructed your track and have successfully displayed it in the Genome Browser, you
may wish to customize the details pages for individual track features. The Genome Browser
automatically creates a default details page for each feature in the track containing the feature's
name, position information, and a link to the corresponding DNA sequence. To view the details page
for a feature in your custom annotation track (in full, pack, or squish display mode), click on the
item's label in the annotation track window.
You can add a link from a details page to an external web page containing additional information about the feature by using the track line url attribute. In the annotation file, set the url attribute in the track line to point to a publicly available page on a web server. The url attribute substitutes each occurrence of '$$' in the URL string with the name defined by the name attribute. You can take advantage of this feature to provide individualized information for each feature in your track by creating HTML anchors that correspond to the feature names in your web page.
Example #4: Here is an example of a file in which the url attribute has been set to point to the file http://genome.ucsc.edu/goldenPath/help/clones.html. The '#$$' appended to the end of the file name in the example points to the HTML NAME tag within the file that matches the name of the feature (cloneA, cloneB, etc.).
browser position chr22:10000000-10020000
browser hide all
track name=clones description="Clones" visibility=2 color=0,128,0 useScore=1 url="http://genome.ucsc.edu/goldenPath/help/clones.html#$$"
#chrom chromStart chromEnd name score
chr22 10000000 10004000 cloneA 960
chr22 10002000 10006000 cloneB 200
chr22 10005000 10009000 cloneC 700
chr22 10006000 10010000 cloneD 600
chr22 10011000 10015000 cloneE 300
chr22 10012000 10017000 cloneF 100 Click here to display this track in the Genome Browser.
Step 6. (Optional) Share your annotation track with others
The previous steps showed you how to upload annotation data for your own use on your own machine.
However, many users would like to share their annotation data with members of their research group
on different machines or with colleagues at other sites. To learn how to make your Genome Browser
annotation track viewable by others, read the section Sharing Your Annotation Track
with Others.
Loading a custom track into the Genome Browser
Using the Genome Browser's custom track upload and management utility, annotation tracks may be added for display in the Genome Browser, deleted from the Genome Browser, or updated with new data and/or display options. You may also use this interface to upload and manage custom track sets for multiple genome assemblies.
To load a custom track into the Genome Browser:
Step 1. Open the Add Custom Tracks page
Select the top blue bar "My Data" menu and click Custom
Tracks. Or, when browsing tracks, click the "add custom tracks" button below the
Genome Browser. (Note: if one or more tracks have already
been uploaded during the current Browser session, additional tracks may be loaded on the Manage
Custom Tracks page. In this case, the button on the Browser page will be labeled "manage
custom tracks" and will automatically direct you to the track management page. See
Displaying and Managing Custom Tracks for more information.)
Step 2. Load the custom track data
The Add Custom Tracks page contains separate sections for uploading custom track data and optional
custom track descriptive documentation. Load the annotation data into the upper section by one of
the following methods:
- Enter one or more URLs for custom tracks (one per line) in the data text box. The Genome Browser supports both the HTTP and FTP (passive-only) protocols. Data provided by a URL may need to be proceeded by a separate line defining type=<track_type> required for some tracks, for example such as "track type=broadPeak".
- Click the "Choose File" button directly above the data text box, then choose a custom track file from your local computer. # or type the pathname of the file into the "upload" text box adjacent to the "Browse" button. The custom track data may be compressed by any of the following programs: gzip (.gz), compress (.Z), or bzip2 (.bz2). Files containing compressed data must include the appropriate suffix in their names.
- Paste or type the custom track data directly into the data box. Because the text in this box will not be saved to a file, this method is not recommended unless you have a copy of the data elsewhere.
Multiple custom tracks may be uploaded at one time on the Add Custom Tracks page through one of the following methods:
- Put all the tracks into the same file (rather than separate files), then load the file via the "Choose File" button.
- Place your track files in a web-accessible location on your server, then load them into the Genome Browser by pasting their URLs into the text input box.
NOTE: Please limit the number of custom tracks that you upload and maintain to less than 1000 tracks. If you have more than this suggested limit of 1000 tracks, please consider setting up a track hub instead.
Step 3. (Optional) Load the custom track description page
If desired, you can provide optional descriptive text (in plain or HTML format) to accompany your
custom track. This text will be displayed when a user clicks the track's description button on the
Genome Browser annotation tracks page. Descriptive text may be loaded by one of the following
methods:
- Click the "Choose File" button directly above the documentation text box, then choose a text file from your local computer.
- Paste or type the custom track data directly into the text input box. Note that the text in this box will not be saved to a file; therefore, this method is not recommended except for temporary documentation purposes.
- If your descriptive text is located on a website, you can reference it from your custom track file by defining the track line attribute "htmlUrl": htmlUrl=<external_url>. In this case, there is no need to insert anything into the documentation text box.
To format your description page in a style that is consistent with standard Genome Browser tracks, click the template link below the documentation text box for an HTML template that may be copied and pasted into a file for editing.
If you load multiple custom tracks simultaneously using one of the methods described in Step 2, a track description can be associated only with the last custom track loaded, unless you upload the descriptive text using the track line "htmlUrl" attribute described above.
Step 4. Upload the track
Click the Submit button to load your custom track data and documentation into the Genome
Browser. If the track uploads successfully, you will be directed to the custom track management page
where you can display your track, update an uploaded track, add more tracks, or delete uploaded
tracks. If the Genome Browser encounters a problem while loading your track, it will display an
error. See the section Troubleshooting Annotation Display Problems for help
in diagnosing custom track problems.
NOTE: Please limit the number of custom tracks that you upload and maintain to less than 1000 tracks. If you have more than this suggested limit of 1000 tracks, please consider setting up a track hub instead.
Displaying and managing custom tracks
After a custom track has been successfully loaded into the Genome Browser, you can display it -- as well as manage your entire custom track set -- via the options on the Manage Custom Tracks page. This page automatically displays when a track has been uploaded into the Genome Browser (see Loading a Custom Track into the Genome Browser). Alternatively, you can access the track management page by clicking the "manage custom tracks" button on the Gateway or Genome Browser annotation tracks pages. (Note that the track management page is available only if at least one track has been loaded during the current browser session; otherwise, this button is labeled "add custom tracks" and opens the Add Custom Track page.)
The table on the Manage Custom Tracks page shows the current set of uploaded custom tracks for the genome and assembly specified at the top of the page. If tracks have been loaded for more than one genome assembly, pulldown lists are displayed; to view the uploaded tracks for a different assembly, select the desired genome and assembly option from the lists.
The following track information is displayed in the Manage Custom Tracks table:
- Name: a hyperlink to the Update Custom Track page where you can update your track configuration and data.
- Description: the value of the "description" attribute from the track line, if present. If no description is included in the input file, this field contains the track name.
- Type: the track type, determined by the Browser based on the format of the data.
- Doc: displays "Y" (Yes) if a description page has been uploaded for the track; otherwise the field is blank.
- Items: the number of data items in the custom track file. An item count is not displayed for tracks lacking individual items (e.g. wiggle format data).
- Pos: the default chromosomal position defined by the track file in either the browser line "position" attribute or the first data line. Click this link to open the Genome Browser or Table Browser at the specified position (Note: only the chromosome name is shown in this column). The Pos column remains blank if the track lacks individual items (e.g. wiggle format data) and the browser line "position" attribute hasn't been set.
Displaying a custom track in the Genome Browser
Click the "go" button to display the entire custom track set for the
specified genome assembly in the Genome Browser. By default, the browser will open to the position
specified in the browser line "position" attribute or first data line of the first custom
track in the table, or the last-accessed Genome Browser position if the track is in wiggle data
format. To open the display at the default position for another track in the list, click the track's
position link in the Pos column.
Viewing a custom track in the Table Browser
Select "Table Browser" from the drop down menu and click the "go" button to
access the data for the custom track set in the Table Browser. The custom tracks will be listed
in the "Custom Tracks" group pulldown list.
Loading additional custom tracks
To load a new custom track into the currently displayed track set, click the "add custom
tracks" button. To change the genome assembly to which the track should be added, select the
appropriate options from the pulldown lists at the top of the page. For instructions on adding a
custom track on the Add Custom Tracks page, see Loading a Custom Track into the
Genome Browser.
Removing one or more custom tracks
To remove custom tracks from the uploaded track set, click the checkboxes in the "delete"
column for all tracks you wish to remove, then click the "delete" button. A custom track
may also be removed by clicking the "Remove custom track" button on the track's
description page. Note: removing the track from the Genome Browser does not delete the track file
from your server or local disk.
Updating a custom track
To update the stored information for a loaded custom track, click the track's link in the
"Name" column in the Manage Custom Tracks table. A custom track may also be updated by
clicking the "Update custom track" button on the track's description page.
The Update Custom Track page provides sections for modifying the track configuration information (the browser lines and track lines), the annotation data, and the descriptive documentation that accompanies the track. Existing track configuration lines are displayed in the top "Edit configuration" text box. In the current implementation of this utility, the existing annotation data is not displayed. Because of this, the data cannot be incrementally edited through this interface, but instead must be fully replaced using one of the data entry methods described in Loading a Custom Track into the Genome Browser. If description text has been uploaded for the track, it will be displayed in the track documentation edit box, where it may be edited or completely replaced. Once you have completed your updates, click the Submit button to upload the new data into the Genome Browser.
If the data or description text for your custom track was originally loaded from a file on your hard disk or server, you should first edit the file, then reload it from the Update Custom Track page using the "Choose File" button. Note that edits made on this page to description text uploaded from a file will not be saved to the original file on your computer or server. Because of this, we recommend that you use the documentation edit box only for changes made to text that was typed or pasted in.
Browser lines
Browser lines are optional, but they give you control of many aspects of the overall display of the Genome Browser window when your annotation file is uploaded. Each line defines one display attribute. Browser lines are in the format:
browser attribute_name attribute_value(s)
For example, if the browser line browser position chr22:1-20000 is included in the
annotation file, the Genome Browser window will initially display the first 20000 bases of chr
22.
The following browser line attribute name/value options are available. The value track_primary_table_name must be set to the name of the primary table on which the track is based. To identify this table, open up the Table Browser, select the correct genome assembly, then select the track name from the track list. The table list will show the primary table. Alternatively, the primary table name can be obtained from a mouseover on the track name in the track control section. You can also find instructions on how to find this table name in the video "How do I learn which tables belong to a data track on the UCSC Genome Browser?".
Note that composite track subtracks are not valid track_primary_table_name values. To find the symbolic name of a composite track, look in the tableName field in the trackDb table, or mouse over the track name in the track control section. It is not possible to display only a subset of the subtracks at this time.
Definition: <track_primary_table_name(s)>. You can find the primary table name by clicking "View Table Schema" from the track's description page, or from the Table Browser. It will be listed as the Primary Table. Alternatively, you can mouse-over the track label in the Browser and look at the URL the link points to. The part after the g= in the URL is the track's primary table name (e.g., for UCSC Genes you will see g=knownGene in the URL. The track primary table is knownGene).
- position <position> - Determines the part of the genome that the Genome Browser will initially open to, in chromosome:start-end format.
- hide all - Hides all annotation tracks except for those listed in the custom track file.
- hide <track_primary_table_name(s)> - Hides the listed tracks. Multiple track names should be space-separated.
- dense all - Displays all tracks in dense mode. NOTE: Use the "all" option cautiously. If the browser display includes a large number of tracks or a large position range, this option may overload your browser's resources and cause an error or timeout.
- dense <track_primary_table_name(s)> - Displays the specified tracks in dense mode. Symbolic names must be used. Multiple track names should be space-separated.
- pack all - Displays all tracks in pack mode. See NOTE for "dense all".
- pack <track_primary_table_name(s)> - Displays the specified tracks in pack mode. Symbolic names must be used. Multiple track names should be space-separated.
- squish all - Displays all tracks in squish mode. See NOTE for "dense all".
- squish <track_primary_table_name(s)> - Displays the specified tracks in squish mode. Symbolic names must be used. Multiple track names should be space-separated.
- full all - Displays all tracks in full mode. See NOTE for "dense all".
- full <track_primary_table_name(s)> - Displays the specified tracks in full mode. Symbolic names must be used. Multiple track names should be space-separated.
Note that the Genome Browser will open to the range defined in the Gateway page search term box or the position saved as the default unless the browser line position attribute is defined in the annotation file. Although this attribute is optional, it's recommended that you set this value in your annotation file to ensure that the track will appear in the display range when it is uploaded into the Genome Browser.
Track lines
Track lines define the display attributes for all lines in an annotation data set. If more than one
data set is included in the annotation file, each group of data must be preceded by a track line
that describes the display characteristics for that set of data. A track line begins with the word
track, followed by one or more attribute=value pairs. Unlike browser
lines - in which each attribute is defined on a separate line - all of the track attributes for a
given set of data are listed on one line with no line breaks. The inadvertent insertion of
a line break into a track line will generate an error when you attempt to upload the annotation
track into the Genome Browser.
The following track line attribute=value pairs are defined in the Genome Browser:
- name=<track_label> - Defines the track label that will be displayed to the left of the track in the Genome Browser window, and also the label of the track control at the bottom of the screen. The name can consist of up to 15 characters, and must be enclosed in quotes if the text contains spaces. We recommend that the track_label be restricted to alpha-numeric characters and spaces to avoid potential parsing problems. The default value is "User Track".
- description=<center_label> - Defines the center label of the track in the Genome Browser window. The description can consist of up to 60 characters, and must be enclosed in quotes if the text contains spaces. The default value is "User Supplied Track".
- type=<track_type> - Defines the track type. The track type attribute is required for BAM, BED detail, bedGraph, bigBarChart, bigBed, bigChain, bigGenePred, bigInteract, bigNarrowPeak, bigMaf, bigPsl, bigWig, broadPeak, CRAM, interact, narrowPeak, Microarray, VCF and WIG tracks.
- visibility=<display_mode> - Defines the initial display mode of the annotation track. Values for display_mode include: 0 - hide, 1 - dense, 2 - full, 3 - pack, and 4 - squish. The numerical values or the words can be used, i.e., full mode may be specified by "2" or "full". The default is "1".
- color=<RRR,GGG,BBB> - Defines the main color for the annotation track. The track color consists of three comma-separated RGB values from 0-255. The default value is 0,0,0 (black).
- itemRgb=On - If this attribute is present and is set to "On", the Genome Browser will use the RGB value shown in the itemRgb field in each data line of the associated BED track to determine the display color of the data on that line.
- colorByStrand=<RRR,GGG,BBB RRR,GGG,BBB> - Sets colors for + and - strands, in that order. The colors consist of three comma-separated RGB values from 0-255 each. The default is 0,0,0 0,0,0 (both black).
-
useScore=<use_score> - If this attribute is present and is set to 1,
the score field in each of the track's data lines will be used to determine the level of
shading in which the data is displayed. The track will display in shades of gray unless the
color attribute is set to 100,50,0 (shades of brown) or 0,60,120 (shades of blue). The
default setting for useScore is "0". This table shows the Genome Browser's
translation of BED score values into shades of gray:
shade score in range ≤ 166 167-277 278-388 389-499 500-611 612-722 723-833 834-944 ≥ 945
-
group=<group> - Defines the annotation track group in which the custom
track will display in the Genome Browser window. By default, group is set to
"user", which causes custom tracks to display at the top of the track listing in the
group "Custom Tracks". The value for "group" must be the
"name" of one of the predefined track groups. To get a list of allowable group names for
an assembly, go to the table browser and select "group: All Tables" "table:
grp" and "get output" where entries in the "name" column may be used
and some examples include "genes","regulation" and "rna".
(Note that mirrors may define other group names in the grp table and that the group setting does not work in track hubs, but does apply for assembly hubs.) - priority=<priority> - When the group attribute is set, defines the display position of the track relative to other tracks within the same group in the Genome Browser window. If group is not set, the priority attribute defines the track's order relative to other custom tracks displayed in the default group, "user".
- db=<UCSC_assembly_name> - When set, indicates the specific genome assembly for which the annotation data is intended; the custom track manager will display an error if a user attempts to load the track onto a different assembly. Any valid UCSC assembly ID may be used (eg. hg18, mm8, felCat1, etc.). The default setting is blank, allowing the custom track to be displayed on any assembly.
- offset=<offset> - Defines a number to be added to all coordinates in the annotation track. The default is "0".
- maxItems=<#> - Defines the maximum number of items the track can contain. The default value is 250. Be aware that tracks with an extremely large number of items can cause system instability. The Kent source utility bedItemOverlapCount can assist in analyzing base overlap with large tracks.
- url=<external_url> - Defines a URL for an external link associated with this track. This URL will be used in the details page for the track. Any "$$"in this string this will be substituted with the item name. There is no default for this attribute.
- htmlUrl=<external_url> - Defines a URL for an HTML description page to be displayed with this track. There is no default for this attribute. A template for a standard format HTML track description is here.
- bigDataUrl=<external_url> - Defines a URL to the data file for BAM, CRAM, bigBed, bigWig or VCF tracks. This is a required attribute for those track types. There is no default for this attribute.
- doWiggle=<on> - The doWiggle setting enables BAM custom tracks to be displayed as bar graphs where the height is proportional to the number of reads mapped to each genomic position. This display is described as a density graph and is best viewed with the track set to full and scaling set to auto-scale. See the Track Database Definition doWiggle entry for more information about the use in hubs and the configuring BAM tracks and the configuring graph-based tracks help pages to learn how to further change the display.
Here is an example of a properly formatted track line using the bigBed format, with accompanying browser line:
browser position chr21:33,031,597-33,041,570
track type=bigBed name="bigBed Example One" description="A bigBed file" bigDataUrl=http://genome.ucsc.edu/goldenPath/help/examples/bigBedExample.bbSharing your annotation track with others
To make your Genome Browser annotation track viewable by people on other machines or at other sites, follow the steps below.
Step 1. Put your formatted annotation file on your web site. Be sure that the file permissions allow it to be read by others.
Step 2. Construct a URL that will link this annotation file to the Genome Browser. The URL must contain 3 pieces of information specific to your annotation data:
-
The species or genome assembly on which your annotation data is based. To automatically display
the most recent assembly for a given organism, set the org parameter: e.g.
org=human. To specify a particular genome assembly for an organism, use the db parameter,db=<databaseName>, where databaseName is the UCSC code for the genome assembly. For a list of these codes, see the Genome Browser FAQ. Examples of this include: db=hg38 (Human Dec. 2013 assembly = GRCh38), db=mm10 (Mouse Dec. 2011 assembly = GRCm38). - Additionally, particular genome assemblies, including assemblies in assembly hubs, can be specified using
the genome parameter,
genome=<databaseName>, which acts in the same manner as the db parameter. Here is an example using the house mouse (Mus musculus, 129S1_SvImJ) assembly hub: http://genome.ucsc.edu/cgi-bin/hgTracks?genome=129S1_SvImJ&hubUrl=http://hgdownload.soe.ucsc.edu/hubs/mouseStrains/hub.txt -
The genome position to which the Genome Browser should initially open. This information is of the
form
position=<chrPosition>, where chrPosition is a chromosome number, with or without a set of coordinates. Examples of this include: position=chr22, position=chr22:15916196-31832390. -
The URL of the annotation file on your web site. This information is of the form
hgt.customText=<url>orhgct_customText=<url>, where url points to the annotation file on your website. An example of an annotation file URL is http://genome.ucsc.edu/goldenPath/help/test.bed.
A special blog post discusses and provides examples of many of these parameters such as
hgct_customText=<url>, db=<databaseName>,
hubUrl=<url>, and genome=<databaseName> to attach
custom tracks, track hubs, assembly hubs, and even track hubs to assembly hubs, all in a
single URL. Read an overview of ways to share Genome Browser data views in the
Sharing Data with Sessions and URLs blog post.
You can add other optional parameters to the URL: (Note: Display may vary if you have conflicting cart variables, for example having both hide all and highlight features.)
-
guidelines=on/off- activate or deactivate the blue guidelines - example link to switch off blue guidelines -
hgFind.matches=<listOfNames>- highlight features given their names - example link to highlight two transcripts of the ABO gene -
hgt.reset=1- show only the default tracks - example link -
hgt.toggleRevCmplDisp=1- show the reverse-complement - example link to show the reverse-complement of the ABO gene -
hgt.labelWidth=<number>- set the size of the left-side label area - example link to increase the label area to 50 characters -
hideTracks=1- hide all tracks - example link to show no tracks at all -
hideTracks=1&<trackName>=full|dense|pack|hide- hide all tracks and show other tracks - example link to show only the Chromosome Bands track and nothing else -
highlight=<db>.<chrom>:<chromStart>-<chromEnd>#<color>|...- highlight one or more regions in a given color on the image. Note that the arguments have to be URL-encoded for Internet browsers, so ":" becomes "%3A", "#" becomes "%23" and "|" becomes "%7"C. - example link to highlight two parts of the ABO locus in red and blue. -
ignoreCookie=1- do not load the user's existing settings saved in the internet browser's UCSC Genome Browser cookie. This means that the link will show the Genome Browser default settings such as track selections, custom tracks, and track hubs. Any changes you make in this new session will, however, affect the user's settings. E.g., if you add a track in this new window, and come back to the genome browser later, the track will still be there. This setting is useful if a website wants to link to the Genome Browser, starting with a "clean slate" but believes the user will come back to the Genome Browser expecting the changes to still be there. -
oligoMatch=pack&hgt.oligoMatch=<dnaSeq>- switch on the Short Match track and highlight a matching sequence - example link to highlight the TATAWAR motif in the ABO locus -
pix=<number>- set the width of the image in pixels - example link to create a 300-pixel wide image -
textSize=<number>- set the size of text font - example link to increase the text font size to 12 pixels -
<trackName>=full|pack|dense|hide- show your current tracks, adding a track and set it to full, pack or dense visibility or hide it, respectively - example link to show the Chromosome Bands track set to "pack" and added to your view as saved in your cart. Please note that for this feature to work with custom tracks you must use the unique name and identifier numberct_name_####assigned by our system. You can determine the name for a custom track using the url, https://genome.ucsc.edu/cgi-bin/cartDump. -
<trackName>.heightPer=<number>- sets the height of the a bigWig track in pixels - example link to set umap bigWig track height to 100 pixels -
<trackName>_hideKids=1- hides a specific super track's individual tracks - example link to hide the Encode Regulation super track -
<trackName>_sel=1- selects specific subtrack to be 'checked', allowing display - example link to select the checkbox for UCSC RefSeq subtrack in the refSeq composite track, allowing display alongside default tracks
If a login and password is required to access data loaded through a URL (e.g., via https: protocol), this information can be included in the URL using the format protocol://user:password@server.com/somepath. Only Basic Authentication is supported for HTTP. Note that passwords included in URLs are not protected. If a password contains a non-alphanumeric character, such as $, the character must be replaced by the hexadecimal representation for that character. For example, in the password mypwd$wk, the $ character should be replaced by %24, resulting in the modified password mypwd%24wk.
For integration into your own website e.g. in an html IFRAME, you can obtain the track image only, without the rest of the genome browser user interface, by replacing hgTracks in the URL with hgRenderTracks, such as in this example:
http://genome.ucsc.edu/cgi-bin/hgRenderTracks?db=hg19&position=chr9%3A136130563-136150630Combine the above pieces of information into a URL of the following format (the information specific to your annotation file is highlighted):
http://genome.ucsc.edu/cgi-bin/hgTracks?org=<organismName>&position=<chrPosition>&hgt.customText=<url>
Example #5:
The following URL will open up the Genome Browser window to display chr22 of the latest human
genome assembly and will show the annotation track pointed to by the URL
http://genome.ucsc.edu/goldenPath/help/test.bed:
http://genome.ucsc.edu/cgi-bin/hgTracks?org=human&position=chr22&hgt.customText=http://genome.ucsc.edu/goldenPath/help/test.bedStep 3. Provide the URL to others. To upload a custom annotation track from a URL into the Genome Browser, paste the URL into the large text edit box on the Add Custom Tracks page, then click the Submit button.
If you'd like to share your annotation track with a broader audience, send the URL for your track—along with a description of the format, methods, and data used—to the UCSC Genome mailing list genome@soe.ucsc.edu.
Example #6:
If you would like to share a URL that your colleague can click on directly, rather than loading it
in the Custom Track tool (as in Example #5), then the URL will need a few extra parameters. Let's
assume that your data is on a server at your institution in one of the large data formats:
bigBed,
bigWig,
bigPsl,
bigBarChart,
bigChain,
bigInteract,
bigGenePred,
bigMaf,
bigNarrowPeak,
BAM,
CRAM, or
VCF.
In this case, the URL must include an hgct_customText parameter, which
simulates the text box on the Custom Tracks page. Also, the URL must include
the bigDataUrl that points to the data file on your server.
So, a clickable URL that opens a remote bigBed track for the hg18 assembly to a certain location on
chr21 would look like this:
http://genome.ucsc.edu/cgi-bin/hgTracks?db=hg18&position=chr21:33038447-33041505&hgct_customText
=track%20type=bigBed%20name=myBigBedTrack%20description=%22a%20bigBed%20track%22%20visibility=
full%20bigDataUrl=http://genome.ucsc.edu/goldenPath/help/examples/bigBedExample.bb
Custom Tracks can also be shared with others through named sessions. First, upload your tracks as discussed in the Loading a Custom Track into the Genome Browser section. Then create a named session that includes your custom tracks by navigating to the "Sessions" page through the "My Data" section in the menu bar. Once there, follow the instructions in the Creating a Session section of the Sessions help page. Once you have saved your custom track into a named session, you can share that session with others by sharing the URL from the "Browser" link or emailing it to them directly by clicking the "Email" link.
Tip: Multiple tracks can be placed into one custom track submission. To do so, create a new file that contains the track lines to each file that will be included. To submit this custom set of tracks, merely use the URL to this new file.
Troubleshooting annotation display problems
Occasionally users encounter problems when uploading annotation files to the Genome Browser. In most cases, these problems are caused by errors in the format of the annotation file and can be tracked down using the information displayed in the error message. This section contains suggestions for resolving common display problems. If you are still unable to successfully display your data, please contact genome@soe.ucsc.edu for further assistance. Messages sent to this address will be posted to the moderated genome mailing list, which is archived on a SEARCHABLE, PUBLIC Google Groups forum.
Problem: When I try one of your examples by cutting and pasting it into
the Genome Browser, I get an error message.
Solution: Check that none of the browser lines, track lines, or data lines
in your annotation file contains a line break. If the example contains GFF or GTF data lines, check
that all the fields are tab-separated rather than space-separated.
Problem: When I click the submit button, I get the error message "line 1 of custom input:".
Solution: Check that none of the browser lines, track lines, or data
lines in your annotation file contains a line break. A common source for this problem is the track
line: all of the attribute pairs must on the same line and must not be separated by a line break.
If you are uploading your annotation file by pasting it into the text box on the Genome Browser
Gateway page, check that the cut-and-paste operation did not inadvertently insert unwanted line
feeds into the longer lines.
Problem: When I click the submit button, I get the error message
"line # of custom input: missing = in var/val pair".
Solution: Check for incorrect syntax in the track lines in the annotation
file. Be sure that each track line attribute pair consists of the format
attribute=attribute name.
Problem: When I click the submit button, I get the error message
"line # of custom input: BED chromStarts[i] must be in ascending order".
Solution: This is most likely caused by a logical conflict in the Genome
Browse software. It accepts custom GFF tracks that have multiple "exons" at the same
position, but not BED tracks. Because the browser translates GFF tracks to BED format before
storing the custom track data, GFF tracks with multiple exons will case an error when the BED is
read back in. To work around this problem, remove duplicate lines in the GFF track.
Problem: When I click the submit button, the Genome Browser
track window displays OK, but my track isn't visible.
Solution: Check the browser and track lines in your annotation file to
make sure that you haven't accidentally set the display mode for the track to hide. If you
are using the Annotation File box on the Genome Browser Gateway page to upload the track, check that
you've entered the correct file name. If neither of these is the cause of the problem, try resetting
the Genome Browser to clear any settings that may be preventing the annotation to display. To reset
the Browser, click the "Reset All User Settings" under the top blue Genome Browser menu. If the
annotation track still doesn't display, you may need to clear the cookies in your Internet browser
as well (refer to your Internet browser's documentation for further information).
Problem: I am trying to upload some custom tracks (.gz files) to the
Genome Browser using a URL from a GEO query. However, the upload is failing with the error
"line 1 of
Solution: The custom track mechanism supports plain BED files (not bigBed)
that are of the type broadPeak or
narrowPeak. Set the track attribute
type=<track_type> to enable the loader to correctly process the
special columns at the end of each line. Your track type entry should consist of two lines: the
first to define the track type and the second to specify the URL. For example:
track type=broadPeak
https://www.ncbi.nlm.nih.gov/geosuppl/...
Problem: I've gotten my annotation track to display, but now I can't make
it go away! How do I remove an annotation track from my Genome Browser display?
Solution: To remove only one track, click the Manage Custom
Tracks button and delete the desired track using the checkbox and Delete button. To quickly
remove all of your custom tracks, reset the Genome Browser to its default settings by clicking on
"Reset All User Settings" under the top blue Genome Browser menu. This reset will also remove any
other customizations you have made to your Genome Browser display.
Problem: I put my custom track files on Dropbox,
Apple iCloud, Google Drive, Amazon Drive, Box.com, Microsoft OneDrive, or
another "online cloud backup" provider and they will not display in the browser.
Why?
OR
Problem: When I try to visualize my custom tracks in the Browser, I
receive the error message "Byte-range request was ignored by server".
Solution: Backup providers used to work for hosting
simple text-based custom tracks, but things have changed. For large custom track data sets, the use
of indexed binary formats such as bigBed and bigWig is preferable and usually fail with backup providers.
These formats provide much faster display performance
because only the portion of the file needed to display the currently viewed region must be
transferred to the Genome Browser server. This requires that they are stored on a real web server.
And to allow this type of display, byte-range support must be
enabled on the web server. No commercial online backup solution supports this,
to our knowledge. To check if your server has byte-range requests enabled,
issue the following command:
curl -I <URL of your file>In order for your server to host bigBed and bigWig files (or track hubs) for Genome Browser display, the command output must contain:
Accept-Ranges: bytesIf you do not receive this output, you may be able to resolve the problem through one of the following actions:
- Store the files on a real web server, e.g. at your University. Check with the systems administrators about the configuration of the server. This will often solve the problem.
- Find another site with a server that supports byte-ranges.
- Install an Apache alternative http server such as Cherokee.
Problem: I used to host files on Dropbox which used to accept byte-range
requests, but I can't get my data to display. Why?
Solution: As described in the previous question, Dropbox is a backup provider
and not a real web server and as such does not support hosting indexed files, even though it used to
work to some extent. Dropbox recently removed their Public Folder feature, which means all links
to files hosted there are inaccessible to the browser. For more information, please refer to this announcement
.
Problem: If I can't host files on backup providers
like Dropbox or Google Drive where can I host my files, especially my bigWigs
and bigBeds?
Solution: The NSF-funded website CyVerse
was created to provide free hosting services to researchers, and allows byte-range
requests, meaning binary files such as BAMs, bigBeds, and bigWigs can be hosted. Please see the
Hosting section of the Track Hub help page for more
information on hosting your data at CyVerse and other alternatives.
Problem: I have a bigBed file with colors in the 9th column. I set the
option itemRgb on in the trackDb file, the track is fine, but why are the boxes all black?
Solution: When type is set to bigBed, the track hub assumes that the
bigBed track is BED3 by default. To allow track hubs to use all fields in a bigBed file, you must
define how many columns to expect. For example, if a bigBed file has nine columns, which would
include an itemRgb field to display a R,G,B color value (e.g. 255,0,0), specify the type
as type bigBed 9 or if the bigBed contains additional non-standard columns, use type bigBed 9 +.
Getting started on Track Hubs
Track hubs are web-accessible directories of genomic data that can be viewed on the UCSC Genome Browser alongside native annotation tracks. Hubs are a useful tool for visualizing a large number of genome-wide data sets. The Track Hub utility allows efficient access to data sets from around the world through the familiar Genome Browser interface. Browser users can display tracks from any public track hub that has been registered with UCSC. Additionally, users can import data from unlisted hubs or can set up, display, and share their own track hubs.
For information on using the Track Hub features, refer to the Genome Browser Track Hub User Guide. For specific information on configuring your trackDb.txt file, refer to the Track Database Definition Document. See also the Basic Hub Quick Start Guide, Quick Start Guide to Organizing Track Hubs into Groupings, Track hub settings blog post, Quick Start Guide to Assembly Hubs and Quick Start Guide to Searchable Track Hubs.
Track Hubs in a single file
Historically, a hub needed a set of text files to specify properties for a track hub and each of the data tracks within the hub. The track hub settings were stored in a three file structure: hub.txt, genomes.txt, and trackDb.txt
myHub/ - directory containing track hub files * hub.txt - a short description of hub properties * genomes.txt - list of genome assemblies included in the hub data * hg19/ - directory of data for the hg19 (GRCh37) human assembly ** trackDb.txt - display properties for tracks in this directory
Now, there is a trackDb option to have your entire track hub inside of one file,
useOneFile on. Adding the useOneFile on line to the hub.txt section of the
file allows the contents of all three files to be referenced inside of one file. We recommend the
single hub.txt file's contents be in the following order: hub.txt, genomes.txt, then trackDb.txt.
hub myExampleHub shortLabel Example Hub longLabel Example Hub for useOneFile option useOneFile on email genome-www@soe.ucsc.edu genome hg19 track vcfExample shortLabel VCF example longLabel VCF: 1000 Genomes phase 1 interim SNVs visibility pack type vcfTabix maxWindowToDraw 200000 bigDataUrl http://genome.ucsc.edu/goldenPath/help/examples/vcfExample.vcf.gz track bamExample shortLabel BAM example longLabel Bam: 1000 Genomes read alignments (individual NA12878) visibility squish type bam pairSearchRange 10000 bamColorMode grey maxWindowToDraw 200000 bigDataUrl http://genome.ucsc.edu/goldenPath/help/examples/bamExample.bam
The hub above can be accessed using the following URL to the single file, singleFileHubExample.txt.
Limitations
Unfortunately, the useOneFile on setting limits the hub to one assembly. The
useOneFile on setting works by having the genomes.txt file point to only one
trackDb.txt file, which is the file itself. Although this method limits you to one genome assembly,
using the setting grants the advantage of not embedding file names in the hub architecture. So a
simple name change to a hub file will no longer require editing the contents inside the hub.txt and
genomes.txt files.
Converting an existing Track Hub to one file
Converting an existing track hub to use the new setting does not require much editing. Simply concatenate the three files into one, with the contents of hub.txt, genomes.txt, and trackDb.txt, in that order. Using the URL to the single file on the My Hubs page will allow you to view your track hub.